10-K: Annual report [Section 13 and 15(d), not S-K Item 405]
Published on June 26, 2025
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM
(MARK ONE)
For the fiscal year ended
OR
For the transition period from ________ to __________
COMMISSION FILE NUMBER
Aethlon Medical, Inc.
(Exact name of registrant as specified in its charter)
| (State or other jurisdiction of | (I.R.S. Employer |
| incorporation or organization) | Identification No.) |
| (Address of principal executive office) | (Zip Code) |
REGISTRANT’S TELEPHONE NUMBER, INCLUDING
AREA CODE: (
SECURITIES REGISTERED PURSUANT TO SECTION 12(b) OF THE EXCHANGE ACT:
|
TITLE OF EACH CLASS |
TRADING SYMBOL |
NAME OF EACH EXCHANGE ON WHICH REGISTERED |
SECURITIES REGISTERED UNDER SECTION 12(g) OF THE EXCHANGE ACT:
NONE
(TITLE OF CLASS)
Indicate by check mark if the registrant is a well-known seasoned issuer,
as defined in Rule 405 of the Securities Act. Yes ☐
Indicate by check mark if the registrant is not required to file reports
pursuant to Section 13 or 15(d) of the Act. Yes ☐
Indicate by check mark whether the registrant
(1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months
(or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements
for the past 90 days.
Indicate by check mark whether the registrant
has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405
of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer”, “smaller reporting company”, and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one)
| Large accelerated filer ☐ | Accelerated filer ☐ | |
| Smaller reporting company |
||
| Emerging growth company |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant
has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial
reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or
issued its audit report.
If securities are registered pursuant to Section
12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction
of an error to previously issued financial statements.
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to § 240.10D-1(b). ☐
Indicate by check mark whether the registrant
is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No
The aggregate market value of the common stock
held by non-affiliates of the registrant as of September 30, 2024 (the last trading day of the registrant’s most recently completed
second quarter) was approximately $
The number of shares of the common stock of the registrant outstanding as of June 24, 2025 was , as adjusted for the Company’s 1-for-8 reverse stock split, which was effective as of the close of business on June 6, 2025 with an effective trading date of June 9, 2025.
TABLE OF CONTENTS
| i |
CAUTIONARY NOTICE REGARDING FORWARD LOOKING STATEMENTS
This Annual Report on Form 10-K, or Annual Report, contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended, or Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act, which are subject to the safe harbor created by those sections.
We may, in some cases, use words such as “anticipate,” “believe,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “will,” “would” or the negative of these terms, and similar expressions that convey uncertainty of future events or outcomes to identify these forward-looking statements. Any statements contained herein that are not statements of historical facts may be deemed to be forward-looking statements and are based upon our current expectations, beliefs, estimates and projections, and various assumptions, many of which, by their nature, are inherently uncertain and beyond our control. Such statements, include, but are not limited to, statements contained in this Annual Report relating to our business, business strategy, products and services we may offer in the future, the timing and results of future regulatory filings, the timing and results of future clinical trials, and capital outlook. Forward-looking statements are based on our current expectations and assumptions regarding our business, the economy and other future conditions. Because forward looking statements relate to the future, they are subject to inherent uncertainties, risks and changes in circumstances that are difficult to predict. Our actual results may differ materially from those contemplated by the forward-looking statements. They are neither statements of historical fact nor guarantees of assurance of future performance. We caution you therefore against relying on any of these forward-looking statements. Important factors that could cause actual results to differ materially from those in the forward looking statements include, but are not limited to, a decline in general economic conditions nationally and internationally; the ability to protect our intellectual property rights; competition from other providers and products; risks in product development; inability to raise capital to fund continuing operations; changes in government regulation; the ability to complete capital raising transactions, and other factors (including the risks contained in Item 1A of this Annual Report under the heading “Risk Factors”) relating to our industry, our operations and results of operations and any businesses that may be acquired by us. Should one or more of these risks or uncertainties materialize, or should the underlying assumptions prove incorrect, actual results may differ significantly from those anticipated, believed, estimated, expected, intended or planned.
Factors or events that could cause our actual results to differ may emerge from time to time, and it is not possible for us to predict all of them, nor can we assess the impact of all factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements we may make. Given these uncertainties, you should not place undue reliance on these forward-looking statements. We cannot guarantee future results, levels of activity, performance or achievements. Except as required by applicable law, we undertake no obligation to and do not intend to update any of the forward-looking statements to conform these statements to actual results.
SUMMARY RISK FACTORS
Below is a summary of the principal factors that make an investment in our securities speculative or risky. This summary does not address all of the risks that we face. Additional discussion of the risks summarized in this risk factor summary, and other risks that we face, can be found below under the heading “Risk Factors” in Part I of this Annual Report and should be carefully considered, together with other information in this Annual Report and our other filings with the SEC before making investment decisions regarding our securities.
| · | We have incurred significant losses and expect to continue to incur losses for the foreseeable future. As a result, our financial statements for the fiscal year ended March 31, 2025 carry a going concern qualification by our independent auditors. | |
| · | We will require additional financing to sustain our operations, achieve our business objectives and satisfy our cash obligations, which may dilute the ownership of our existing stockholders. | |
| · | We have limited experience in identifying and working with large-scale contracts with medical device manufacturers; manufacture of our devices must comply with good manufacturing practices in the United States. | |
| · | Delays, interruptions or the cessation of production by our third-party suppliers of important materials or delays in qualifying new materials has and may continue to prevent or delay our ability to manufacture our Hemopurifier. |
| ii |
| · | Our Hemopurifier technology may become obsolete. | |
| · | If we fail to comply with extensive regulations of U.S. and foreign regulatory agencies, the commercialization of our products could be delayed or prevented entirely. | |
| · | If we are unable to maintain compliance with the listing requirements of the Nasdaq Capital Market, our common stock may be delisted from the Nasdaq Capital Market, which could have a material adverse effect on our financial condition and could make it more difficult for you to sell your shares. | |
| · |
As a public company with limited financial resources undertaking the launch of new medical technologies, we may have difficulty attracting and retaining executive management and directors.
|
|
| · |
We plan to expand our operations, which may strain our resources; our inability to manage our growth could delay or derail implementation of our business objectives.
|
|
| · |
Our success is dependent in part on our executive officers.
|
|
| · | Delays in successfully commencing or completing our planned clinical trials could jeopardize our ability to obtain regulatory approval and sustain our operations. |
| iii |
PART I
ITEM 1. BUSINESS
Unless otherwise indicated or the context otherwise requires, references to the “Company”, “Aethlon”, “we”, “us” and “our” refer to Aethlon Medical, Inc.
Overview and Corporate History
Overview
We are a medical therapeutic company focused on developing the Hemopurifier® (HP), a clinical-stage immunotherapeutic device intended for applications in cancer, life-threatening viral infections, and organ transplantation and other areas of significant unmet needs. In human studies (164 sessions with 38 patients), the Hemopurifier was used safely and demonstrated the potential to remove enveloped viruses. In pre-clinical studies, the Hemopurifier has exhibited the capacity to remove harmful extracellular vesicles (EVs) and enveloped viruses from biological fluids, utilizing its proprietary lectin-based mechanism. These extracellular vesicles have been implicated in disease processes such as immune suppression and metastasis in cancer as well as in the progression of severe life-threatening infectious diseases. The U.S. Food and Drug Administration (“FDA”) has designated the Hemopurifier as a “Breakthrough Device” for two independent indications:
| · | the treatment of individuals with advanced or metastatic cancer who are unresponsive to or intolerant of standard of care therapy, and with cancer types in which extracellular vesicles have been shown to participate in the development or severity of the disease; and | |
| · |
the treatment of life-threatening viruses for which no approved therapies currently exist. |
We are also evaluating the Hemopurifier’s potential in additional clinical contexts based on its mechanism of action and preclinical findings.
Oncology
We believe that the Hemopurifier may be a substantial advancement in the treatment of patients with advanced and metastatic cancer through its design to bind to and remove harmful remove harmful extracellular vesicles particles that promote the growth and spread of tumors. In October 2022, we formed a wholly-owned subsidiary in Australia to initially conduct oncology-related clinical research, then seek regulatory approval and commercialize our Hemopurifier in Australia.
We completed an in vitro binding study of extracellular vesicles from cancer patient samples, to provide pre-clinical evidence to support our trial design and translational endpoints. Our study indicated positive results from this study, providing evidence that our Hemopurifier removes extracellular vesicles, or EVs, from plasma. This translational study provides pre-clinical evidence to support our phase 1 safety, feasibility and dose-finding clinical trials of our Hemopurifier in patients with solid tumors who have stable or progressive disease during anti-PD-1 monotherapy treatment, such as Keytruda® or Opdivo®.
We have launched in an Australia safety, feasibility and dose-finding clinical trials of the Hemopurifier in cancer patients with solid tumors who have stable or progressive disease during anti-PD-1 monotherapy treatment, such as Keytruda® (pembrolizumab) or Opdivo® (nivolumab). The primary endpoint of the approximately nine to 18-patients, is safety. Exploratory analyses will be conducted to explore the number of HP treatments required to produce sustained reductions of EVs as well as improve anti-tumor T cell activity. We plan to open a similarly designed trial in India.
The following three hospitals in Australia have received ethics committee approval, have gone through training on our device and are open for patient enrollment: Royal Adelaide Hospital in Adelaide, Australia and Pindara Private Hospital in the Gold Coast section of Australia and GenesisCare North Shore Hospital in Sydney, Australia. As of June 26, 2025, we have treated three participants in the first of the three treatment cohorts. Once these patients have completed the pre-specified 7-day safety follow-up period, the data will be presented to an independent Data Safety Monitoring Board (DSMB). The DSMB will provide a recommendation to Aethlon senior leadership on advancing to the next cohort where participants will receive 2 HP treatments during the one week treatment period.
| 1 |
The Company continues to pursue approval of a similar clinical trial in India. HREC approval has previously been obtained at Medanta Medicity Hospital. Following this a meeting with Subject Expert Committee (SEC) of the India Regulatory Agency CDSCO was held 5JUN2025. We are awaiting the formal approval letter of the CDSCO. The clinical trial at Medanta can commence following a Site Initiation Visit (SIV) by the company’s India CRO, Qualtran.
Life-Threatening Viral Infections
We also believe that the Hemopurifier can be part of the broad-spectrum treatment of life-threatening highly glycosylated, or carbohydrate coated, viruses that are not addressed with an already approved treatment. In small-scale or early feasibility human studies, the Hemopurifier has been used in the past to treat individuals infected with human immunodeficiency virus, or HIV, hepatitis-C and Ebola.
Additionally, in vitro, the Hemopurifier has been demonstrated to capture Ebola, Marburg virus, Zika, Lassa, MERS-CoV, Cytomegalovirus, Epstein-Barr, Herpes simplex, Chikungunya, Dengue, West Nile, H1N1 swine flu, H5N1 bird flu, and the reconstructed 1918 Spanish flu virus. In several cases, these studies were conducted in collaboration with leading government or non-government research institutes.
The Hemopurifier has previously been studied under FDA and international regulatory frameworks for the treatment of severe SARS-CoV-2 infection. While we terminated our U.S. and India-based COVID-19 studies due to low ICU patient volume and shifting priorities, these programs demonstrated real-world use of the Hemopurifier in critically ill patients. We maintain an open IDE for viral indications to preserve optionality for future outbreaks or emergent pathogens.
We have sufficient inventory of Hemopurifiers to support our ongoing oncology trial in Australia as well as any near-term expansion of that study or potential trial activity in India. While we have received FDA approval to begin manufacturing at our San Diego facility under our IDE supplement, we are still awaiting FDA approval of a separate supplement to qualify an additional supplier of a key Hemopurifier component. We continue to work with the FDA on this process.
Pre-Clinical Exploration of Additional Clinical Uses for the Hemopurifier
The Aethlon R&D laboratory continues to explore potential new indications for the Hemopurifier. We have published in the peer-reviewed journal Transplant Immunology the ability of the device to remove extracellular vesicles and their microRNA cargo from acellular perfusates of discarded kidneys that had undergone normothermic machine perfusion.
On May 12, 2025, the results of our pre-clinical ex vivo study entitled “Ex Vivo Removal of CD41 positive platelet microparticles from Plasma by a Medical Device containing a Galanthus nivalis agglutinin (GNA) affinity resin” were published in the pre-print vehicle bioRxiv. This manuscript has been submitted to a peer-reviewed publication for review.
Platelet-derived extracellular vesicles (PD-EVs) are the most numerous EV population in the body and are released by platelets in response to a variety of stimuli. The cargo contained within these EVs have been noted to take part in damage to blood vessels, activation of immune cells and spread of tumor cells. Excessive levels of PD-EVs have been implicated in a myriad of diseases including cancer, lupus, systemic sclerosis, multiple sclerosis, Alzheimer’s disease, sepsis, acute and Long COVID.
We hypothesized that the Aethlon Hemopurifier which contains a propriety GNA affinity resin would remove platelet derived EVs from plasma. In this experiment two hundred milliliters on donated healthy human plasma were circulated over the Aethlon Hemoupurifier (HP) to simulate a clinical HP session. The study results showed a 98.5% removal of platelet -derived EVs at a timepoint equivalent to a 4-hour HP treatment. The results of this study support the current Australian Clinical Trial in Oncology as well as open the investigation of the Hemopurifier in many indications.
| 2 |
Extracellular vesicles have been implicated in the pathogenesis of Long COVID. As we had previously demonstrated removal of extracellular vesicles by the Hemopurifier in a patient with severe acute COVID-19 infection, we hypothesized that patients with Long COVID would have extracellular vesicles with the mannose sugar on their surface that would bind to the affinity resin in our device. We partnered with investigators at the Univ of California San Francisco Medical Center Long COVID clinic to obtain samples from participants with Long COVID as well as controls that had had COVID -10 infection but had recovered. The data to be presented will review the binding of larger and smaller extracellular vesicles to the GNA lectin and the lectin affinity resin, respectively. We believe the data from this pre-clinical study calls for additional study of the Hemopurifier and look forward to receiving feedback from the Long COVID scientific community at the Keystone Symposium.
Successful outcomes of human trials will also be required by the regulatory agencies of certain foreign countries where we plan to market and sell the Hemopurifier. Some of our patents may expire before FDA approval or approval in a foreign country, if any, is obtained. However, we believe that certain patent applications and/or other patents issued to us more recently will help protect the proprietary nature of our Hemopurifier treatment technology.
In addition to the foregoing, we are monitoring closely the impact of inflation, recent bank failures and the war between Russia and Ukraine and the military conflicts in Israel and the surrounding areas, as well as related political and economic responses and counter-responses by various global factors on our business. Given the level of uncertainty regarding the duration and impact of these events on capital markets and the U.S. economy, we are unable to assess the impact on our timelines and future access to capital. The full extent to which inflation, recent bank failures and the ongoing military conflicts will impact our business, results of operations, financial condition, clinical trials and preclinical research will depend on future developments, as well as the economic impact on national and international markets that are highly uncertain.
On March 10, 1999, Aethlon, Inc., a California corporation, Hemex, Inc., a Delaware corporation and the accounting predecessor to Aethlon, Inc., and Bishop Equities, Inc., a publicly traded Nevada corporation, completed an Agreement and Plan of Reorganization structured to result in Bishop Equities, Inc.’s acquisition of all of the outstanding common stock of Aethlon, Inc. and Hemex, Inc. Under the plan’s terms, Bishop Equities, Inc. issued shares of its common stock to the stockholders of Aethlon, Inc. and Hemex, Inc. such that Bishop Equities, Inc. then owned 100% of each company. Upon completion of the transaction, Bishop Equities, Inc. was renamed Aethlon Medical, Inc. Our executive offices are located at 11555 Sorrento Valley Road, Suite 203, San Diego, California 92121. Our telephone number is (619) 941-0360. Our website address is www.aethlonmedical.com. The information contained on, or that can be accessed through, our website is not part of, and is not incorporated into, this Annual Report.
The Mechanism of Action (MOA) of the Hemopurifier
The Hemopurifier is a lectin-affinity plasmapheresis extracorporeal device designed for the removal of harmful extracellular vesicles and life-threatening enveloped viruses from the plasma component of the bloodstream. In the United States, the Hemopurifier is classified as a combination product whose regulatory jurisdiction is the Center for Devices and Radiological Health, or CDRH, the branch of FDA responsible for the premarket approval of all medical devices.
In our current applications, our Hemopurifier can be used with approved dialysis machines serving as a blood pump. It could also potentially be developed as part of a proprietary closed system with its own pump and tubing set, negating the requirement for dialysis infrastructure.
The Hemopurifier - Clinical Experience
Hepatitis C and HIV
The initial clinical development of the Hemopurifier focused on the viral infections Hepatitis C and HIV. Clinical trials conducted in India and a safety trial demonstrated the removal of both viruses from the bloodstream with a benign safety profile. Prior to FDA approval of the IDE feasibility study, we conducted investigational HCV treatment studies at the Apollo Hospital, Fortis Hospital, and the Medanta Medicity Institute in India. In the Medanta Medicity Institute study, 12 HCV-infected individuals were enrolled to receive three six-hour Hemopurifier treatments during the first three days of a 48-week peginterferon+ribavirin treatment regimen. The study was conducted under the leadership of Dr. Vijay Kher. Dr. Kher’s staff reported that Hemopurifier therapy was well tolerated and without device-related adverse events in the 12 patients treated.
| 3 |
Of these 12 patients, ten completed the Hemopurifier-peginterferon+ribavirin treatment protocol, including eight genotype-1 patients and two genotype-3 patients. Eight of the ten patients achieved a sustained virologic response, which is the clinical definition of treatment cure and is defined as undetectable HCV in the blood 24 weeks after the completion of the 48-week peginterferon+ribavirin drug regimen. Both genotype-3 patients achieved a sustained virologic response, while six of the eight genotype-1 patients achieved a sustained virologic response, which defines a cure of the infection. Our IDE safety study in end stage renal disease patients on dialysis who were infected with HCV was conducted at DaVita MedCenter Dialysis in Houston, Texas. We reported that there were no device-related adverse events in enrolled subjects who met the study inclusion-exclusion criteria. We also reported that an average capture of 154 million copies of HCV (in International Units, I.U.) within the Hemopurifier during four-hour treatments.
The initial clinical development of the Hemopurifier was focused on the viral infections Hepatitis C and HIV. Clinical trials conducted in India and a safety trial demonstrated the removal of both viruses from the bloodstream with a benign safety profile. Prior to FDA approval of the IDE feasibility study, we conducted investigational HCV treatment studies at the Apollo Hospital, Fortis Hospital, and the Medanta Medicity Institute in India. In the Medanta Medicity Institute study, 12 HCV-infected individuals were enrolled to receive three six-hour Hemopurifier treatments during the first three days of a 48-week peginterferon+ribavirin treatment regimen. The study was conducted under the leadership of Dr. Vijay Kher. Dr. Kher’s staff reported that Hemopurifier therapy was well tolerated and without device-related adverse events in the 12 treated.
Of these 12 patients, ten completed the Hemopurifier-peginterferon+ribavirin treatment protocol, including eight genotype-1 patients and two genotype-3 patients. Eight of the ten patients achieved a sustained virologic response, which is the clinical definition of treatment cure and is defined as undetectable HCV in the blood 24 weeks after the completion of the 48-week peginterferon+ribavirin drug regimen. Both genotype-3 patients achieved a sustained virologic response, while six of the eight genotype-1 patients achieved a sustained virologic response, which defines a cure of the infection. Our IDE safety study in end stage renal disease patients on dialysis who were infected with HCV was conducted at DaVita MedCenter Dialysis in Houston, Texas. We reported that there were no device-related adverse events in enrolled subjects who met the study inclusion-exclusion criteria. We also reported that an average capture of 154 million copies of HCV (in International Units, I.U.) within the Hemopurifier during four-hour treatments.
In addition to treating Ebola and HCV-infected individuals, we also conducted a single proof-of-principle treatment study at the Sigma New Life Hospital in an AIDS patient who was not being administered HIV antiviral drugs. In the study, viral load was reduced by 93% as the result of 12 Hemopurifier treatments (each four hours in duration) that were administered over the course of one month.
With the advent of highly effective anti-retroviral drugs for HIV (HAART), and curative direct acting antivirals (DACs) for Hepatitis C, clinical development for these indications was abandoned.
Ebola Virus-Single Patient Emergency Use
Under Emergency use conditions a single patient with Ebola infection with multiple organ dysfunction was treated with the Hemopurifier at Frankfurt University Hospital in Germany. The patient tolerated a single 6.5-hour Hemopurifier treatment. Prior to treatment, the Ebola viral load was measured at 400,000 copies/ml. The post-treatment viral load was 1,000 copies/ml. Calculations by the treating physician indicated that 242 million copies of Ebola virus were captured within the Hemopurifier during treatment. The patient made a full recovery. Based on this experience, the Company filed an Expanded Access protocol with the FDA to treat Ebola virus infected patients in up to ten centers in the United States and a corresponding protocol was approved by HealthCanada. These protocols remain open, allowing Hemopurifier treatment to be offered to patients presenting for care in both countries. In 2018, the FDA designated the Hemopurifier as a Breakthrough Device “… for the treatment of life-threatening viruses that are not addressed with approved therapies.”
Severe Acute SARS-CoV-2/COVID-19 Infection – Emergency Use and Clinical Trials
SARS-COV-2, the causative agent of COVID-19 is a member of the coronavirus family, which includes the original SARS virus, SARS-CoV, and the MERS virus. SARS-CoV-2, found to contain mannose on the envelope surface. This suggests that the Hemopurifier could potentially clear it from biological fluids, including blood.
| 4 |
Under Single Patient Emergency Use regulations, we have treated two patients with COVID-19 with the Hemopurifier. We published a manuscript reviewing case studies covering those two Single Patient Emergency Use treatments entitled “Removal of COVID-19 Spike Protein, Whole Virus, Exosomes and Exosomal microRNAs by the Hemopurifier® Lectin-Affinity Cartridge in Critically Ill Patients with COVID-19 Infection” in the peer-reviewed journal Frontiers in Medicine
The manuscript described the use of the Hemopurifier for a total of nine sessions in two critically ill COVID-19 patients. The first case study demonstrated the improvement in the patient who was a SARS-COV-2 positive COVID-19 present at entry to the hospital, with associated coagulopathy, or CAC, lung injury, inflammation, and tissue injury despite the absence of demonstrable COVID-19 viremia at the start of treatment at Day 22.This patient received eight Hemopurifier treatments without complications and eventually was weaned from a ventilator and was discharged from the hospital. Plasma samples from this patient revealed a decrease in extracellular vesicle counts over the course of the eight treatments and decreases in exosomal microRNAs associated with the development of coagulopathy and acute lung injury.
The second patient case study demonstrated in vivo removal of SARS-CoV-2 virus from the blood stream of an infected patient. This patient completed a six-hour Hemopurifier treatment without complications and subsequently was placed on continuous renal replacement therapy, or CRRT. The patient ultimately expired three hours after being placed on CRRT because of the advanced stage of the patient’s disease.
On June 17, 2020, the FDA approved a supplement to our open IDE for the Hemopurifier in viral disease to allow for the testing of the Hemopurifier in patients with SARS-CoV-2/COVID-19 in a New Feasibility Study. That study was designed to enroll up to 40 subjects at up to 20 centers in the United States. Subjects had to have an established laboratory diagnosis of COVID-19, be admitted to an ICU, and have acute lung injury and/or severe or life-threatening disease, among other criteria. Endpoints for this study, in addition to safety, include reduction in circulating virus, as well as clinical outcomes (NCT # 04595903). In June 2022, the Company completed the treatment protocol for its first patient in this study.
In June 2022, the Company completed the treatment protocol of the only participant enrolled in the study. study. The patient received one HP treatment daily for 4 days. This patient died following cardiac arrest (not related to the HP treatment) as a consequence of severe COVID-19 pneumonia. Blood samples taken from the patient did not reveal any evidence of viremia. Plasma sent for cytokine analysis revealed a numeric decrease in the levels of IP-10, MCP-1, and IL-10.
A similarly designed trial was also conducted in India. One patient was enrolled on February 16, 2022, at Medanta Medicity Hospital, Gurugram, Haryana 12200, India. The patient tolerated one HP treatment daily for three days. On 19 February 2022, in the first 15 min during the 3rd treatment, one nonserious Grade 2 AE was reported (hemolysis and leaking of the filter). The filter was replaced, and therapy resumed without sequalae. On Day #4 the patient suffered asystole and died due to clinical deterioration unrelated to the device. During the first Hemopurifier treatment (T1) there was a gradual decrease in viral load from the baseline at 4923 copies/mL decreasing steadily to 1307 copies/mL over five hours, indicating a 73% reduction from baseline. At the beginning of the second Hemopurifier treatment (T2), the viral load was 850 copies/mL, dropped below the lower limit of quantification within an hour, and remained undetectable, suggesting rapid clearance. The viral load before the third treatment (T3) was below the quantification limit but unexpectedly rose at 3 hours (636 copies/mL), peaking at 4 hours (1583 copies/mL), and slightly decreasing at 5 hours (1104 copies/mL). This irregular pattern suggests possible delayed RNA release, sample variability, or another biological factor affecting detection. The cumulative data shows a reduced SARs-CoV-2 viral load during the first two Hemopurifier treatments but not during the third treatment.
Due to lack of eligible patients in the ICU the clinical trial was closed as November 22, 2022.
Oncology- U.S. Clinical Trial in Head and Neck Cancer
A single center clinical trial entitled “Depleting Exosomes to Improve Response to Immune Therapy in Head and Neck Squamous Cell Cancer: An Early Feasibility Phase I Clinical Trial” was conducted under a US IDE at the University of Pittsburgh. This was a single arm Phase 1 clinical trial designed to evaluate the safety and efficacy of the Hemopurifier plus pembrolizumab for the treatment of patients with recurrent or metastatic head and neck squamous cell cancer. All patients were treated with pembrolizumab every 21 days as standard of care. The patients were to receive a 4-hour Hemopurifier treatment before Pembrolizumab infusions 2 occasions 21 days apart. A total of 2 patients were enrolled in the study with the first occurring on Dec 14, 2020. The first patients received 2 HP treatments, and the second patient received one HP treatment. The second treatment in the second patient was terminated due to operator error. Eighteen no serious adverse events occurred in the two patients with none thought related to the device.
| 5 |
The only exploratory efficacy laboratory analysis that was performed in this study was a determination of the total nanoparticle concentrations in the 1st patient prior to and for 14 days after the second HP treatment. Total nanoparticle concentrations decreased following each Hemopurifier treatment. Following Hemopurifier treatment, the total nanoparticle concentrations rose by about Day 7 but did not reach the baseline levels. Exosomes levels are a component of the total nanoparticle concentration but exosome levels over time were not specifically determined.
Research and Development Costs
A substantial portion of our operating budget is used for research and development activities. The cost of research and development, all of which has been charged to operations, amounted to approximately $2,212,000 and $2,520,000 in the fiscal years ended March 31, 2025 and 2024, respectively.
Recent Developments
On June 25, 2025, the Company received notice from Nasdaq stating the Company has regained compliance with Listing Rule 5550(a)(2), and that the matter is now closed.
Reverse Split – Following the approval of a reverse stock split at a Special Meeting of Stockholders on May 13, 2025, our Board of Directors approved a 1-for-8 reverse stock split of our outstanding shares of Common Stock, effective as of the close of business on June 6, 2025 with an effective trading date of June 9, 2025. Accordingly, each eight shares of outstanding common stock held by stockholders were combined into one share of common stock. Our authorized common stock remained at 60,000,000 shares following the stock split. We issued an additional 77 shares as a result of rounding up fractional shares related to the reverse stock split.
On June 2, 2025 a second patient was treated with the Hemopurifier at GenesisCare North Shore Hospital in Sydney, Australia. The patient was treated with the Aethlon Hemopurifier for 4 hours in a single day and tolerated the procedure without complications. The patient will have follow-up safety visits, EV and T cell measurements as well as imaging for clinical response.
Intellectual Property
We currently own or have license rights to a number of U.S. and foreign patents and patent applications and endeavor to continually improve our intellectual property position. We consider the protection of our technology, whether owned or licensed, to the exclusion of use by others, to be vital to our business. While we intend to focus primarily on patented or patentable technology, we also rely on trade secrets, unpatented property, know-how, regulatory exclusivity, patent extensions and continuing technological innovation to develop our competitive position. We also own certain trademarks.
Our success depends in large part on our ability to protect our proprietary technology, including the Hemopurifier product platform, and to operate without infringing the proprietary rights of third parties. We rely on a combination of patent, trade secret, copyright and trademark laws, as well as confidentiality agreements, licensing agreements and other agreements, to establish and protect our proprietary rights. Our success also depends, in part, on our ability to avoid infringing patents issued to others. If we were judicially determined to be infringing on any third-party patent, we could be required to pay damages, alter our products or processes, obtain licenses or cease sales of products or certain activities.
To protect our proprietary medical technologies, including the Hemopurifier product platform and other scientific discoveries, we have a portfolio of over 32 issued patents and pending applications worldwide. We currently have three issued U.S. patents and 14 issued patents in countries outside of the United States. In addition, we have 15 patent applications pending worldwide related to our Hemopurifier product platform and other technologies. We are seeking additional patents on our scientific discoveries.
It is possible that our pending patent applications may not result in issued patents, that we will not develop additional proprietary products that are patentable, that any patents issued to us may not provide us with competitive advantages or will be challenged by third parties and that the patents of others may prevent the commercialization of products incorporating our technology. Furthermore, others may independently develop similar products, duplicate our products or design around our patents. U.S. patent applications are not immediately made public, so it is possible that a third party may obtain a patent on a technology we are actively using.
| 6 |
There is a risk that any patent applications that we file and any patents that we hold or later obtain could be challenged by third parties and declared invalid or unenforceable. For many of our pending applications, patent interference proceedings may be instituted with the U.S. Patent and Trademark Office, or the USPTO, when more than one person files a patent application covering the same technology, or if someone wishes to challenge the validity of an issued patent. At the completion of the interference proceeding, the USPTO will determine which competing applicant is entitled to the patent, or whether an issued patent is valid. Patent interference proceedings are complex, highly contested legal proceedings, and the USPTO’s decision is subject to appeal. This means that if an interference proceeding arises with respect to any of our patent applications, we may experience significant expenses and delays in obtaining a patent, and if the outcome of the proceeding is unfavorable to us, the patent could be issued to a competitor rather than to us. Third parties can file post-grant proceedings in the USPTO, seeking to have issued patent invalidated, within nine months of issuance. This means that patents undergoing post-grant proceedings may be lost, or some or all claims may require amendment or cancellation, if the outcome of the proceedings is unfavorable to us. Post-grant proceedings are complex and could result in a reduction or loss of patent rights. The institution of post-grant proceedings against our patents could also result in significant expenses.
Patent law outside the United States is uncertain and in many countries, is currently undergoing review and revisions. The laws of some countries may not protect our proprietary rights to the same extent as the laws of the United States. Third parties may attempt to oppose the issuance of patents to us in foreign countries by initiating opposition proceedings. Opposition proceedings against any of our patent filings in a foreign country could have an adverse effect on our corresponding patents that are issued or pending in the United States. It may be necessary or useful for us to participate in proceedings to determine the validity of our patents or our competitors’ patents that have been issued in countries other than the United States. This could result in substantial costs, divert our efforts and attention from other aspects of our business, and could have a material adverse effect on our results of operations and financial condition. Outside of the United States, we currently have pending patent applications or issued patents in Europe, India, Russia, Canada, Japan, Singapore and Hong Kong.
In addition to patent protection, we rely on unpatented trade secrets and proprietary technological expertise. It is possible that others could independently develop or otherwise acquire substantially equivalent technology, somehow gain access to our trade secrets and proprietary technological expertise or disclose such trade secrets, or that we may not successfully ultimately protect our rights to such unpatented trade secrets and proprietary technological expertise. We rely, in part, on confidentiality agreements with our marketing partners, employees, advisors, vendors and consultants to protect our trade secrets and proprietary technological expertise. We cannot assure you that these agreements will not be breached, that we will have adequate remedies for any breach or that our unpatented trade secrets and proprietary technological expertise will not otherwise become known or be independently discovered by competitors.
Patents
The following table lists our issued patents and patent applications, including their ownership status, including relevant patent term adjustments (PTA), which is a process of extending the term of a U.S. patent:
Patents Issued in the United States
| PATENT # | PATENT NAME |
ISSUANCE DATE |
OWNED OR LICENSED |
EXPIRATION DATE |
| 9,707,333 | Extracorporeal removal of microvesicular particles | 7/18/17 | Owned | 1/6/29 |
| 9,364,601 | Extracorporeal removal of microvesicular particles | 6/14/16 | Owned | 5/30/29 |
| 8,288,172 | Extracorporeal removal of microvesicular particles | 10/16/12 | Owned | 3/09/27 05/30/29 (with 813 days Patent Term Adjustment (PTA)) |
| 7 |
Patent Applications Pending in the United States
| APPLICATION # | APPLICATION NAME |
FILING DATE |
OWNED OR LICENSED |
| 17/918,085 | Devices and methods for treating a coronavirus infection and symptoms thereof | 10/10/22 | Owned |
| 18/700571 | Devices and methods for treating a viral infection and symptoms thereof | 04/11/24 | Owned |
Foreign Patents
| PATENT # | PATENT NAME |
ISSUANCE DATE |
OWNED OR LICENSED |
EXPIRATION DATE |
| 60 2011 035 500.7 | Methods for quantifying exosomes (Germany) | 3/01/17 | Owned | 7/07/31 |
| 2591359 | Methods for quantifying exosomes (France) | 3/01/17 | Owned | 7/07/31 |
| 2591359 | Methods for quantifying exosomes (Great Britain) | 3/01/17 | Owned | 7/07/31 |
| 11804372 | Methods for quantifying exosomes (Spain) | 3/01/17 | Owned | 7/07/31 |
| 2644855 | Extracorporeal removal of microvesicular particles (Canada) | 11/19/19 | Owned | 3/09/27 |
| 502019000055563 | Extracorporeal removal of microvesicular particles (Germany) | 4/24/19 | Owned | 3/09/27 |
| 1993600 | Extracorporeal removal of microvesicular particles (Switzerland) | 4/24/19 | Owned | 3/09/27 |
| 1993600 | Extracorporeal removal of microvesicular particles (Spain) | 4/24/19 | Owned | 3/09/27 |
| 1993600 | Extracorporeal removal of microvesicular particles (France) | 4/24/19 | Owned | 3/09/27 |
| 1993600 | Extracorporeal removal of microvesicular particles (Great Britain) | 4/24/19 | Owned | 3/09/27 |
| 502019000055563 | Extracorporeal removal of microvesicular particles (Italy) | 4/24/19 | Owned | 3/09/27 |
| 1993600 | Extracorporeal removal of microvesicular particles (Netherlands) | 4/24/19 | Owned | 3/09/27 |
| 1993600 | Extracorporeal removal of microvesicular particles (Sweden) | 4/24/19 | Owned | 3/09/27 |
| 1126138 | Extracorporeal removal of microvesicular particles (Hong Kong) | 6/19/20 | Owned | 3/09/27 |
| 8 |
Pending Foreign Patent Applications
| APPLICATION # | APPLICATION NAME | FILING DATE |
OWNED OR LICENSED |
| 8139/DELNP/2008 | Extracorporeal removal of microvesicular particles (exosomes) (India) | 3/9/07 | Owned |
| 2021256402 | Devices and methods for treating a coronavirus infection and symptoms thereof (Australia) | 10/16/22 | Owned |
| 3178687 | Devices and methods for treating a coronavirus infection and symptoms thereof (Canada) | 9/29/22 | Owned |
| 21788894.0 | Devices and methods for treating a coronavirus infection and symptoms thereof (Europe) | 10/26/22 | Owned |
| 62023077768.7 | Devices and methods for treating a coronavirus infection and symptoms thereof (Hong Kong) | 08/17/23 | Owned |
| 297109 | Devices and methods for treating a coronavirus infection and symptoms thereof (Israel) | 10/6/22 | Owned |
| 2023-505809 | Devices and methods for treating a coronavirus infection and symptoms thereof (Japan) | 10/12/22 | Owned |
| 2022361924 | Devices and methods for treating a viral infection and symptoms thereof (Australia) | 04/12/24 | Owned |
| 2024-522200 | Devices and methods for treating a viral infection and symptoms thereof (Japan) | 04/12/24 | Owned |
| 3235306 | Devices and methods for treating a viral infection and symptoms thereof (Canada) | 4/11/2024 | Owned |
| 22881946.2 | Devices and methods for treating a viral infection and symptoms thereof (Europe) | 4/23/2024 | Owned |
| 62025103640 | Devices and methods for treating a viral infection and symptoms thereof (Hong Kong) | 2/18/2025 | Owned |
Pending International Patent Applications
| APPLICATION # | APPLICATION NAME |
FILING DATE |
OWNED OR LICENSED |
| PCT/US2024/015614 | Removal of exosomes, ectosomes, mirnas, circulating nucleic acids, and viral particles with | 2/13/24 | Owned |
Trademarks
| APPLICATION NAME | Countries | Priority Date | OWNED OR LICENSED |
| *SANSAGITTA | Madrid, Australia, Canada, the EU, UK, and India | 7/8/2021 | Owned |
* The US Application for SANSAGITTA abandoned on 12/2/24. It was used as the basis application for a Madrid registration, and the corresponding above-listed designated country registrations can be converted to national applications to avoid abandonment.
| 9 |
Trademarks
In addition to the Sansagitta trademarks noted in the above table, we also have trademark registrations in the United States for Hemopurifier and Aethlon Medical, Inc., and obtained a trademark registration in India for Hemopurifier. We also have common law trademark rights in Aethlon ADAPT™ and ELLSA™.
Industry & Competition
The industry for treating infectious disease and cancer is extremely competitive, and companies developing new treatment procedures face significant capital and regulatory challenges. As our Hemopurifier is a clinical-stage device, we have the additional challenge of establishing medical industry support, which will be driven by treatment data resulting from human clinical studies. Should our device become market cleared by the FDA or the regulatory body of another country, we may face significant competition from well-funded pharmaceutical organizations. Additionally, we would likely need to establish large-scale production of our device in order to be competitive. Our competitors include blood filters produced by ExThera Medical Corporation.
Government Regulation
The Hemopurifier is subject to regulation by numerous regulatory bodies, primarily the FDA, and comparable international regulatory agencies. These agencies require manufacturers of medical devices to comply with applicable laws and regulations governing the development, testing, manufacturing, labeling, marketing, storage, distribution, advertising and promotion, and post-marketing surveillance reporting of medical devices. As the primary mode of action of the Hemopurifier is attributable to the device component of this combination product, the CDRH has primary jurisdiction over its premarket development, review and approval. Failure to comply with applicable requirements may subject a device and/or its manufacturer to a variety of administrative sanctions, such as issuance of warning letters, import detentions, civil monetary penalties and/or judicial sanctions, such as product seizures, injunctions and criminal prosecution.
FDA’s Pre-market Clearance and Approval Requirements
Each medical device we seek to commercially distribute in the United States will require either a prior 510(k) clearance, unless it is exempt, or a pre-market approval from the FDA. Generally, if a new device has a predicate that is already on the market under a 510(k) clearance, the FDA will allow that new device to be marketed under a 510(k) clearance; otherwise, a premarket approval, or PMA, is required. Medical devices are classified into one of three classes—Class I, Class II or Class III—depending on the degree of risk associated with each medical device and the extent of control needed to provide reasonable assurance of safety and effectiveness. Class I devices are deemed to be low risk and are subject to the general controls of the Federal Food, Drug and Cosmetic Act, such as provisions that relate to: adulteration; misbranding; registration and listing; notification, including repair, replacement, or refund; records and reports; and good manufacturing practices. Most Class I devices are classified as exempt from pre-market notification under section 510(k) of the FD&C Act, and therefore may be commercially distributed without obtaining 510(k) clearance from the FDA. Class II devices are subject to both general controls and special controls to provide reasonable assurance of safety and effectiveness. Special controls include performance standards, post market surveillance, patient registries and guidance documents. A manufacturer may be required to submit to the FDA a pre-market notification requesting permission to commercially distribute some Class II devices. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a previously cleared 510(k) device, are placed in Class III. A Class III device cannot be marketed in the United States unless the FDA approves the device after submission of a PMA. However, there are some Class III devices for which FDA has not yet called for a PMA. For these devices, the manufacturer must submit a pre-market notification and obtain 510(k) clearance in orders to commercially distribute these devices. The FDA can also impose sales, marketing or other restrictions on devices in order to assure that they are used in a safe and effective manner. We believe that the Hemopurifier will be classified as a Class III device and as such will be subject to PMA submission and approval.
Pre-market Approval Pathway
A pre-market approval application must be submitted to the FDA for Class III devices for which the FDA has required a PMA. The pre-market approval application process is much more demanding than the 510(k) pre-market notification process. A pre-market approval application must be supported by extensive data, including but not limited to technical, preclinical, clinical trials, manufacturing and labeling to demonstrate to the FDA’s satisfaction reasonable evidence of safety and effectiveness of the device.
| 10 |
After a pre-market approval application is submitted, the FDA has 45 days to determine whether the application is sufficiently complete to permit a substantive review and thus whether the FDA will file the application for review. The FDA has 180 days to review a filed pre-market approval application, although the review of an application generally occurs over a significantly longer period of time and can take up to several years. During this review period, the FDA may request additional information or clarification of the information already provided. Also, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device.
Although the FDA is not bound by the advisory panel decision, the panel’s recommendations are important to the FDA’s overall decision making process. In addition, the FDA may conduct a preapproval inspection of the manufacturing facility to ensure compliance with the Quality System Regulation, or QSR. The agency also may inspect one or more clinical sites to assure compliance with FDA’s regulations.
Upon completion of the PMA review, the FDA may: (i) approve the PMA which authorizes commercial marketing with specific prescribing information for one or more indications, which can be more limited than those originally sought; (ii) issue an approvable letter which indicates the FDA’s belief that the PMA is approvable and states what additional information the FDA requires, or the post-approval commitments that must be agreed to prior to approval; (iii) issue a not approvable letter which outlines steps required for approval, but which are typically more onerous than those in an approvable letter, and may require additional clinical trials that are often expensive and time consuming and can delay approval for months or even years; or (iv) deny the application. If the FDA issues an approvable or not approvable letter, the applicant has 180 days to respond, after which the FDA’s review clock is reset.
Emergency Use Authorizations, or EUAs, are granted by FDA in public health emergencies but allow use of the authorized device only during the period of the respective public health emergency, and do not change the requirement to ultimately seek PMA approval after the authorization period has ended.
Clinical Trials
Clinical trials are almost always required to support pre-market approval and are sometimes required for 510(k) clearance. In the United States, for significant risk devices, these trials require submission of an application for an IDE to the FDA. The IDE application must be supported by appropriate data, such as animal and laboratory testing results, showing it is safe to test the device in humans and that the testing protocol is scientifically sound. The IDE must be approved in advance by the FDA for a specific number of patients at specified study sites. During the trial, the sponsor must comply with the FDA’s IDE requirements for investigator selection, trial monitoring, reporting and recordkeeping. The investigators must obtain patient informed consent, rigorously follow the investigational plan and study protocol, control the disposition of investigational devices and comply with all reporting and recordkeeping requirements. Clinical trials for significant risk devices may not begin until the IDE application is approved by the FDA and the appropriate institutional review boards, or IRBs, at the clinical trial sites. An IRB is an appropriately constituted group that has been formally designated to review and monitor medical research involving subjects and which has the authority to approve, require modifications in, or disapprove research to protect the rights, safety and welfare of human research subjects. The FDA or the IRB at each site at which a clinical trial is being performed may withdraw approval of a clinical trial at any time for various reasons, including a belief that the risks to study subjects outweigh the benefits or a failure to comply with FDA or IRB requirements. Even if a trial is completed, the results of clinical testing may not demonstrate the safety and effectiveness of the device, may be equivocal or may otherwise not be sufficient to obtain approval or clearance of the product.
Ongoing Regulation by the FDA
Even after a device receives clearance or approval and is placed on the market, numerous regulatory requirements apply. These include:
| · | establishment registration and device listing; | |
| · | the QSR, which requires manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the manufacturing process; | |
| · | labeling regulations and the FDA prohibitions against the promotion of products for uncleared, unapproved or “off-label” uses and other requirements related to promotional activities; |
| 11 |
| · | medical device reporting regulations, which require that manufactures report to the FDA if their device may have caused or contributed to a death or serious injury, or if their device malfunctioned and the device or a similar device marketed by the manufacturer would be likely to cause or contribute to a death or serious injury if the malfunction were to recur; | |
| · | corrections and removal reporting regulations, which require that manufactures report to the FDA field corrections or removals if undertaken to reduce a risk to health posed by a device or to remedy a violation of the FDCA that may present a risk to health; and | |
| · | post market surveillance regulations, which apply to certain Class II or III devices when necessary to protect the public health or to provide additional safety and effectiveness data for the device. |
Some changes to an approved PMA device, including changes in indications, labeling or manufacturing processes or facilities, require submission and FDA approval of a new PMA or PMA supplement, as appropriate, before the change can be implemented. Supplements to a PMA often require the submission of the same type of information required for an original PMA, except that the supplement is generally limited to that information needed to support the proposed change from the device covered by the original PMA. The FDA uses the same procedures and actions in reviewing PMA supplements as it does in reviewing original PMAs.
Failure by us or by our suppliers to comply with applicable regulatory requirements can result in enforcement action by the FDA or state authorities, which may include any of the following sanctions:
| · | warning or untitled letters, fines, injunctions, consent decrees and civil penalties; | |
| · | customer notifications, voluntary or mandatory recall or seizure of our products; | |
| · | operating restrictions, partial suspension or total shutdown of production; | |
| · | delay in processing submissions or applications for new products or modifications to existing products; | |
| · | withdrawing approvals that have already been granted; and | |
| · | criminal prosecution. |
The Medical Device Reporting laws and regulations require us to provide information to the FDA when we receive or otherwise become aware of information that reasonably suggests our device may have caused or contributed to a death or serious injury as well as a device malfunction that likely would cause or contribute to death or serious injury if the malfunction were to recur. In addition, the FDA prohibits an approved device from being marketed for off-label use. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including substantial monetary penalties and criminal prosecution.
Newly discovered or developed safety or effectiveness data may require changes to a product’s labeling, including the addition of new warnings and contraindications, and also may require the implementation of other risk management measures. Also, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory clearance or approval of our products under development.
| 12 |
Healthcare Regulation
In addition to the FDA’s restrictions on marketing of pharmaceutical products, the U.S. healthcare laws and regulations that may affect our ability to operate include: the federal fraud and abuse laws, including the federal anti-kickback and false claims laws; federal data privacy and security laws; and federal transparency laws related to payments and/or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and other healthcare professionals (such as physicians assistants and nurse practitioners) and teaching hospitals. Many states have similar laws and regulations that may differ from each other and federal law in significant ways, thus complicating compliance efforts. For example, states have anti-kickback and false claims laws that may be broader in scope than analogous federal laws and may apply regardless of payor. In addition, state data privacy laws that protect the security of health information may differ from each other and may not be preempted by federal law. Moreover, several states have enacted legislation requiring pharmaceutical manufacturers to, among other things, establish marketing compliance programs, file periodic reports with the state, make periodic public disclosures on sales and marketing activities, report information related to drug pricing, require the registration of sales representatives, and prohibit certain other sales and marketing practices. These laws may adversely affect our sales, marketing and other activities with respect to any product candidate for which we receive approval to market in the United States by imposing administrative and compliance burdens on us.
Because of the breadth of these laws and the narrowness of available statutory exceptions and regulatory safe harbors, it is possible that some of our business activities, particularly any sales and marketing activities after a product candidate has been approved for marketing in the United States, could be subject to legal challenge and enforcement actions. If our operations are found to be in violation of any of the federal and state laws described above or any other governmental regulations that apply to us, we may be subject to significant civil, criminal, and administrative penalties, including, without limitation, damages, fines, imprisonment, exclusion from participation in government healthcare programs, additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the regulatory approval, manufacture and marketing of regulated products or the reimbursement thereof. For example, in the United States, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, ACA, among other things, reduced and/or limited Medicare reimbursement to certain providers and imposed an annual excise tax of 2.3% on any entity that manufactures or imports medical devices offered for sale in the United States, with limited exceptions. However, the 2020 federal spending package permanently eliminated, effective January 1, 2020, this ACA-mandated medical device tax. On June 17, 2021, the U.S. Supreme Court dismissed a challenge on procedural grounds that argued the ACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. Further, on August 16, 2022, President Biden signed the Inflation Reduction Act of 2022, or IRA, into law, which among other things, extends enhanced subsidies for individuals purchasing health insurance coverage in ACA marketplaces through plan year 2025. The IRA also eliminates the "donut hole" under the Medicare Part D program beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost and creating a new manufacturer discount program. It is possible that the ACA will be subject to judicial or congressional challenges in the future. It is unclear how such challenges and any additional healthcare reform measures will impact the ACA.
| 13 |
Other legislative changes have been proposed and adopted since the ACA was enacted. The Budget Control Act of 2011, as amended by subsequent legislation, further reduces Medicare’s payments to providers by two percent through fiscal year 2032. These reductions may reduce providers’ revenues or profits, which could affect their ability to purchase new technologies. Furthermore, the healthcare industry in the United States has experienced a trend toward cost containment as government and private insurers seek to control healthcare costs by imposing lower payment rates and negotiating reduced contract rates with service providers. In July 2021, the Biden Administration released an executive order, “Promoting Competition in the American Economy,” which contained provisions relating to prescription drugs. On September 9, 2021, in response to this executive order, the U.S. Department of Health and Human Services, or HHS, released a Comprehensive Plan for Addressing High Drug Prices that outlines principles for drug pricing reform and sets out a variety of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can take to advance these principles. Further, the IRA, among other things (i) directs HHS to negotiate the price of certain high-expenditure, single-source drugs and biologics covered under Medicare and (ii) imposes rebates under Medicare Part B and Medicare Part D to penalize price increases that outpace inflation. These provisions will take effect progressively starting in fiscal year 2023, although they may be subject to legal challenges. HHS has and will continue to issue and update guidance as these programs are implemented. It is currently unclear how the IRA will be implemented but is likely to have a significant impact on the pharmaceutical industry. In addition, in response to the Biden administration’s October 2022 executive order, on February 14, 2023, HHS released a report outlining three new models for testing by the Center for Medicare and Medicaid Innovation which will be evaluated on their ability to lower the cost of drugs, promote accessibility, and improve quality of care. It is unclear whether the models will be utilized in any health reform measures in the future.
Legislation could be adopted in the future that limits payments for our products from governmental payors. In addition, commercial payors such as insurance companies, could adopt similar policies that limit reimbursement for medical device manufacturers’ products.
Coverage and Reimbursement
In both the U.S. and international markets, the use of medical devices is dependent in part on the availability of reimbursement from third-party payors, such as government and private insurance plans. Healthcare providers that use medical devices generally rely on third-party payors to pay for all or part of the costs and fees associated with the medical procedures being performed or to compensate them for their patient care services. Should our Hemopurifier or any other products under development be approved for commercialization by the FDA, any such products may not be considered cost-effective, reimbursement may not be available in the United States or other countries, if approved, and reimbursement may not be sufficient to allow sales of our future products on a profitable basis. The coverage decisions of third-party payors will be significantly influenced by the assessment of our future products by health technology assessment bodies. If approved for use in the United States, we expect that any products that we develop, including the Hemopurifier, will be purchased primarily by medical institutions, which will in turn bill various third-party payors for the health care services provided to patients at their facility. Payors may include the Centers for Medicare & Medicaid Services, or CMS, which administers the Medicare program and works in partnership with state governments to administer Medicaid, other government programs and private insurance plans. The process involved in applying for coverage and reimbursement from CMS is lengthy and expensive. Further, Medicare coverage is based on our ability to demonstrate that the treatment is “reasonable and necessary” for Medicare beneficiaries. Even if products utilizing our Hemopurifier technology receive FDA and other regulatory clearance or approval, they may not be granted coverage and reimbursement by any payor, including by CMS. Many private payors use coverage decisions and payment amounts determined by CMS as guidelines in setting their coverage and reimbursement policies and amounts. However, no uniform policy for coverage and reimbursement for medical devices exists among third-party payors in the United States. Therefore, coverage and reimbursement can differ significantly from payor to payor.
Manufacturing
Historically, manufacturing of our Hemopurifier was conducted in collaboration with a contract manufacturer based in California, operating under current Good Manufacturing Practice, or cGMP, regulations promulgated by the FDA. Our contract manufacturer is registered with the FDA. To date, production of the Hemopurifier has been limited to quantities necessary to support our clinical studies.
In May 2024, the FDA approved the use of our own manufacturing for the production of Hemopurifiers. We have since initiated manufacturing activities at our facility under cGMP conditions to support ongoing and planned clinical development.
Our costs of compliance with federal, state and local environmental laws have been immaterial to date.
| 14 |
Sources and Availability of Raw Materials and the Names of Principal Suppliers
Aethlon personnel assemble the various components of the Hemopurifier with materials from our various suppliers, which are purchased and released by Aethlon. Specifically, the Hemopurifier contains three critical components with limited available suppliers. The GNA lectin is sourced from Vector Laboratories Inc. and also is available from other suppliers. Our intended transition from Vector Laboratories to a new supplier for GNA is delayed as we work with the FDA for approval of our supplement to our IDE, which is required to make this manufacturing change. The base cartridge on which the Hemopurifier is constructed is sourced from Medica S.p.A and we are dependent on the continued availability of these cartridges. Although there are other suppliers, the process of qualifying a new supplier takes time and regulatory approvals must be obtained. We currently purchase the diatomaceous earth from Janus Scientific, Inc., as the distributor; however, the product is manufactured by Imerys Minerals Ltd. There potentially are other suppliers of this product, but as with the cartridges, qualifying and obtaining required regulatory approvals takes time and resources.
Sales and Marketing
We do not currently have any sales and marketing capability. With respect to commercialization efforts in the future, we intend to build or contract for distribution, sales and marketing capabilities for any product candidate that is approved. From time to time, we have had and are having strategic discussions with potential collaboration partners for our product candidates, although no assurance can be given that we will be able to enter into one or more collaboration agreements for our product candidates on acceptable terms, if at all.
Product Liability
The risk of product liability claims, product recalls and associated adverse publicity is inherent in the testing, manufacturing, marketing and sale of medical products. We have limited clinical trial liability insurance coverage. It is possible that future insurance coverage may not be adequate or available. We may not be able to secure product liability insurance coverage on acceptable terms or at reasonable costs when needed. Any liability for mandatory damages could exceed the amount of our coverage. A successful product liability claim against us could require us to pay a substantial monetary award. Moreover, a product recall could generate substantial negative publicity about our products and business and inhibit or prevent commercialization of other future product candidates.
Employees
As of June 26, 2025, we had 9 full-time employees and no part-time employees. All of our employees are located in the United States. We do intend to hire additional employees. We utilize, whenever appropriate, consultants in order to conserve cash and resources.
We believe our employee relations are good. None of our employees are represented by a labor union or are subject to collective-bargaining agreements.
| 15 |
ITEM 1A. RISK FACTORS
An investment in our securities involves a high degree of risk. You should carefully consider the risks described below as well as the other information in this Annual Report before deciding to invest in or maintain your investment in our company. The risks described below are not intended to be an all-inclusive list of all of the potential risks relating to an investment in our securities. Any of the risk factors described below could significantly and adversely affect our business, prospects, financial condition and results of operations. Additional risks and uncertainties not currently known or that are currently considered to be immaterial may also materially and adversely affect our business. As a result, the trading price or value of our securities could be materially adversely affected and you may lose all or part of your investment.
Risks Relating to Our Financial Position and Need for Additional Capital
We have incurred significant losses and expect to continue to incur losses for the foreseeable future.
We have never been profitable. We did not generate any revenue during the fiscal years ended March 31, 2025 and March 31, 2024. In prior fiscal years we did record revenue from government contracts. We do not currently have any research grants or contracts. It is possible that we may not be able to enter into future government contracts. Future profitability, if any, will require the successful commercialization of our Hemopurifier technology or any other product that we develop or from additional government contract or grant income we may obtain. We may not be able to successfully commercialize the Hemopurifier or any other products, and even if commercialization is successful, we may never be profitable. While we currently have over $5.5 million in cash and cash equivalents and have been carrying out certain expense reductions since November 2023, our planned additional expense reductions may not materialize and/or our patient recruitment may occur more rapidly than expected along with the concomitant increases in expenses; therefore there is substantial doubt that our cash on hand will carry the company for 12 months beyond the filing date of the financial statements included in this Annual Report.
We do plan to access the equity markets for additional capital, however, there can be no assurance that we will be able to access such additional capital.
We will require additional financing to sustain our operations, achieve our business objectives and satisfy our cash obligations, which may dilute the ownership of our existing stockholders.
We will require significant additional financing for our operations and for expected additional future clinical trials in the United States, India and Australia, regulatory clearances, and continued research and development activities for the Hemopurifier and other future products. In addition, as we expand our activities, our overhead costs to support personnel, laboratory materials and infrastructure will increase. We may also choose to raise additional funds in debt or equity financings if they are available to us on reasonable terms to increase our working capital and to strengthen our financial position. Any sale of additional equity or convertible debt securities could result in dilution of the equity interests of our existing stockholders. Additionally, new investors may require that we and certain of our stockholders enter into voting arrangements that give them additional voting control or representation on our Board of Directors. If required financing is unavailable to us on reasonable terms, or at all, we may be unable to support our operations, including our research and development activities, which would have a material adverse effect on our ability to commercialize our products or continue our business.
Our ability to raise additional funds may be adversely impacted by our ability to remain listed on Nasdaq, the potential worsening global economic conditions and disruptions to and volatility in the credit and financial markets in the United States, including due to bank failures, actual or perceived changes in interest rates and economic inflation, and worldwide resulting from macroeconomic factors. Because of the numerous risks and uncertainties associated with product development, we cannot predict the timing or amount of increased expenses and cannot assure you that we will ever be profitable or generate positive cash flow from operating activities.
| 16 |
We may not currently or in the future be able to continue as a going concern.
The financial statements in this Annual Report have been prepared on a going concern basis of accounting, which assumes that we will continue as a going concern, and do not reflect any adjustments that might result if the Company is unable to continue as a going concern. The Company’s ability to continue as a going concern is dependent on our ability to generate revenues and raise capital. To date, we have not generated sufficient revenues to provide cash flows that enable us to finance our operations internally. In connection with an evaluation conducted by our management during the preparation of the financial statements included in this Annual Report, management concluded that there were conditions and events which raised substantial doubt as to the Company’s ability to continue as a going concern within twelve months after the date of the issuance of the financial statements included in this Annual Report.
The uncertainty regarding our ability to continue as a going concern could materially adversely affect our share price and our ability to service our indebtedness, raise new capital or enter into commercial transactions. To address these matters, we may take actions that materially and adversely affect our business, including significant reductions in research, development, administrative and commercial activities, reduction of our employee base, and ultimately curtailing or ceasing operations, any of which could materially adversely affect our business, financial condition, results of operations and share price. In addition, doubts about our ability to continue as a going concern could impact our relationships with partners, vendors and other third parties and our ability to obtain, maintain or renew contracts with them, or negatively impact our negotiating leverage with such parties, which could have a material adverse effect on our business, financial condition and results of operations. Furthermore, any loss of key personnel, employee attrition or material erosion of employee morale arising out of doubts about our ability to operate as a going concern could have a material adverse effect on our ability to effectively conduct our business and could impair our ability to execute our strategy and implement our business objectives, thereby having a material adverse effect on our business, financial condition and results of operations.
Risks Related to Our Business Operations
Delays, interruptions or the cessation of production by our third-party suppliers of important materials or delays in qualifying new materials, has and may continue to prevent or delay our ability to manufacture our Hemopurifier.
Most of the raw materials used in the process for manufacturing our Hemopurifier are available from more than one supplier. However, there are materials within the manufacturing and production process that come from single suppliers. We do not have written contracts with all of our single source suppliers, and at any time they could stop supplying our orders. FDA review of a new supplier is required if these materials become unavailable from our current suppliers. In the recent past, we experienced an interruption in the manufacturing of our Hemopurifier as we sought to transition to a new supplier of galanthus nivalis agglutinin, or GNA, used in the manufacture of our Hemopurifier. We have not received the required FDA approval of our IDE supplement for a new qualified supplier of the GNA and are working with the FDA to gain approval of this supplier. Although we have resumed purchasing GNA from our prior supplier, it is possible that we could experience future disruptions from this supplier as we work to qualify a second supplier. FDA review of the new second supplier could take several additional months to obtain.
In addition, an uncorrected impurity, a supplier’s variation in a raw material or testing, either unknown to us or incompatible with its manufacturing process, or any other problem with our materials, testing or components, could prevent or delay the release of our Hemopurifiers for use in our clinical trials. For example, in late 2020, we identified during our device quality review procedures prior to product release that one of our critical suppliers had produced a Hemopurifier component that was not produced to our specifications, although no affected Hemopurifiers were released into our inventory or to any clinical trial sites. Any such future supplier issues could have a material adverse impact on our business, results of operations and financial condition.
| 17 |
Difficulties in manufacturing our Hemopurifier could have an adverse effect upon our expenses, our product revenues and our ability to complete our clinical trials.
We received approval from the FDA for our IDE supplement to manufacture Hemopurifiers at our site in San Diego. The manufacturing of our Hemopurifier is difficult and complex. To support our current clinical trial needs, we comply with and intend to continue to comply with current Food Manufacturing Practices, or cGMP in the manufacture of our product. Our ability to adequately manufacture and supply our Hemopurifier in a timely matter is dependent on the uninterrupted and efficient operation of our facilities and those of third parties producing raw materials and supplies upon which we rely in our manufacturing. The manufacture of our products may also be impacted by:
| · | availability or contamination of raw materials and components used in the manufacturing process, particularly those for which we have no other source or supplier; | |
| · | our ability to comply with new regulatory requirements, including our ability to comply with cGMP; | |
| · | natural disasters; | |
| · | changes in forecasts of future demand for product components; | |
| · | potential facility contamination by microorganisms or viruses; | |
| · | updating of manufacturing specifications; | |
| · | product quality success rates and yields; and | |
| · | global viruses and pandemics. |
Any future interruption in the manufacture and supply of our Hemopurifier could delay shipments of our Hemopurifier for use in clinical trials in the United States, Australia and India.
Our products are manufactured with raw materials that are sourced from specialty suppliers with limited competitors and we may therefore be unable to access the materials we need to manufacture our products.
Specifically, the Hemopurifier contains three critical components with limited supplier numbers. The base cartridge on which the Hemopurifier is constructed is sourced from Medica S.p.A and we are dependent on the continued availability of these cartridges. We currently purchase the diatomaceous earth from Janus Scientific Inc., our distributor; however, the product is manufactured by Imerys Minerals Ltd., which is the only supplier of this product. The GNA is sourced from Vector Laboratories, Inc. and also is available from other suppliers; however, Sigma Aldrich is our only potential back up supplier at this time and we are in the process of working with the FDA to obtain regulatory approval for this supplier. A business interruption at any of these sources, including the interruption resulting from the delay in obtaining FDA approval of our new GNA supplier, has and may continue to have a material impact on our ability to manufacture the Hemopurifier.
| 18 |
We face intense competition in the medical device industry.
We compete with numerous U.S. and foreign companies in the medical device industry, and many of our competitors have greater financial, personnel, operational and research and development resources than we do. We believe that because the field of exosome research is burgeoning, multiple competitors are or will be developing competing technologies to address exosomes in cancer. Progress is constant in the treatment and prevention of viral diseases, so the opportunities for the Hemopurifier may be reduced there as well. Diagnostic technology may be developed that can supplant diagnostics we are developing for viruses and cancer. Our commercial opportunities will be reduced or eliminated if our competitors develop and market products for any of the diseases we target that:
| · | are more effective; | |
| · | have fewer or less severe adverse side effects; | |
| · | are better tolerated; | |
| · | are more adaptable to various modes of dosing; | |
| · | are easier to administer; or | |
| · | are less expensive than the products or product candidates we are developing. |
Even if we are successful in developing the Hemopurifier and obtain FDA and other regulatory approvals necessary for commercialization, our products may not compete effectively with other successful products. Researchers are continually learning more about diseases, which may lead to new technologies for treatment. Our competitors may succeed in developing and marketing products that are either more effective than those that we may develop, alone or with our collaborators, or that are marketed before any products we develop are marketed. Our competitors include fully integrated pharmaceutical companies and biotechnology companies as well as universities and public and private research institutions. Many of the organizations competing with us have substantially greater capital resources, larger research and development staffs and facilities, greater experience in product development and in obtaining regulatory approvals, and greater marketing capabilities than we do. If our competitors develop more effective pharmaceutical treatments for infectious disease or cancer, or bring those treatments to market before we can commercialize the Hemopurifier for such uses, we may be unable to obtain any market traction for our products, or the diseases we seek to treat may be substantially addressed by competing treatments. If we are unable to successfully compete against larger companies in the pharmaceutical industry, we may never generate significant revenue or be profitable.
We have limited experience in identifying and working with large-scale contracts with medical device manufacturers; manufacture of our devices must comply with good manufacturing practices in the United States.
To achieve the levels of production necessary to commercialize our Hemopurifier and any other future products, we will need to secure large-scale manufacturing agreements with contract manufacturers which comply with good manufacturing practice standards and other standards prescribed by various federal, state and local regulatory agencies in the United States and any other country of use. We have limited experience coordinating and overseeing the manufacture of medical device products on a large-scale. It is possible that manufacturing and control problems will arise as we attempt to commercialize our products and that manufacturing may not be completed in a timely manner or at a commercially reasonable cost. In addition, we may not be able to adequately finance the manufacture and distribution of our products on terms acceptable to us, if at all. If we cannot successfully oversee and finance the manufacture of our products if they obtain regulatory clearances, we may never generate revenue from product sales and we may never be profitable.
| 19 |
We have in the past experienced a material weakness in our internal controls over financial reporting. If we fail to maintain effective internal controls and fail to remediate any future or present control deficiencies, our ability to produce accurate and timely financial statements could be impaired, which could harm our operating results, our ability to operate our business and our reputation with investors, ultimately leading to a decline in the price of our Common Stock.
As a public company, we are subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, the Sarbanes-Oxley Act, and the rules and regulations of the applicable listing standards of Nasdaq. In particular, Section 404 of the Sarbanes-Oxley Act requires that we evaluate and determine the effectiveness of our internal controls over financial reporting. It also requires our independent registered public accounting firm to attest to our evaluation of our internal controls over financial reporting.
As disclosed in Item 9A in our Annual Report on Form 10-K for the fiscal year ended March 31, 2024, management identified a material weakness in the segregation of duties within our financial systems. Specifically, user access controls were not sufficiently maintained to properly restrict both user and privileged access to financial applications within our accounting software system to initiate, record and approve entries. We also noted that check stock was secured in an authorized signatory’s office. During 2017 through 2020, the Company incorrectly recorded accrued commission liability of approximately $404,000. The Company reversed accrued commission liability of approximately $404,000 during the year ended March 31, 2024 related to this error in accounting under U.S. GAAP. The Company originally failed to correctly apply appropriate accounting principles in recording the transaction, and the error was not detected and corrected in a timely manner, resulting in an adjustment to the financial statements. Management has discussed with counsel appropriate measures to record such potential commission liabilities in the future and will implement a quarterly review of all accruals. The reversal of the accrued commission liability into equity as of March 31, 2024 corrected the impact of the error.
Since that time, we have implemented several remediation measures, including enhanced user access controls, segregation of duties, relocation of check stock to a secure, access-controlled area, and the implementation of a quarterly review process for all significant accruals. Management has also consulted with legal counsel to clarify how potential commission liabilities should be recorded in the future. The reversal of the commission accrual into equity as of March 31, 2024 corrected the impact of the historical error.
As of March 31, 2025, management has concluded that the previously identified material weakness has been remediated. While we are committed to maintaining a robust control environment, there can be no assurance that future material weaknesses will not be identified.
If we have difficulty maintaining effective internal controls over financial reporting, or if we identify a material weakness in our internal controls over financial reporting in the future, we may not detect errors on a timely basis, such that it could harm our operating results, adversely affect our reputation, cause our stock price to decline, or result in inaccurate financial reporting or material misstatements in our annual or interim financial statements. We may be unable to maintain compliance with securities laws, stock exchange listing requirements and debt instruments’ covenants regarding the timely filing of accurate periodic reports, which could lead to investigations by Nasdaq, the SEC or other regulatory authorities or litigations with our creditors and/or stockholders, hence requiring additional management attention and impairing our ability to operate our business. Our liquidity, access to capital markets and perceptions of our creditworthiness may be adversely affected. We could be required to implement expensive and time-consuming remedial measures. Our independent registered public accounting firm may issue reports that are adverse in the event it is not satisfied with the level at which our internal control over financial reporting is documented, designed, or operating, or if it is not satisfied with our remediation of any identified material weaknesses. Any failure to maintain effective disclosure controls and internal control over financial reporting could have a material adverse effect on our business, financial position, results of operations, and cash flows.
| 20 |
Our Hemopurifier technology may become obsolete.
Our Hemopurifier product may be made unmarketable prior to commercialization by us by new scientific or technological developments by others with new treatment modalities that are more efficacious and/or more economical than our products. The homeland security industry is growing rapidly with many competitors that are trying to develop products or vaccines to protect against infectious disease. Any one of our competitors could develop a more effective product which would render our technology obsolete. Further, our ability to achieve significant and sustained penetration of our key target markets will depend upon our success in developing or acquiring technologies developed by other companies, either independently, through joint ventures or through acquisitions. If we fail to develop or acquire, and manufacture and sell, products that satisfy our customers’ demands, or we fail to respond effectively to new product announcements by our competitors by quickly introducing competitive products, then market acceptance of our products could be reduced and our business could be adversely affected. Our products may not remain competitive with products based on new technologies.
We are highly dependent on our key personnel, and if we are not successful in attracting and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
Our ability to compete in the highly competitive biotechnology and medical device industries depends upon our ability to attract and retain highly qualified managerial, scientific, and medical personnel. We are highly dependent on our management, scientific, and medical personnel. The loss of the services of any of our executive officers or other key employees and our inability to find suitable replacements could potentially harm our business, prospects, financial condition or results of operations.
We do not currently carry key man life insurance policies on any of our key executive officers which would assist us in recouping our costs in the event of the loss of those officers. If any of our key officers were to leave us, it could make it impossible, if not cause substantial delays and costs, to implement our long-term business objectives and growth.
Our inability to attract and retain qualified personnel could impede our ability to achieve our business objectives.
We have 9 full-time employees. We utilize, whenever appropriate, consultants in order to conserve cash and resources. Although we believe that these employees and consultants will be able to handle most of our additional administrative, research and development and business development in the near term, we will nevertheless be required over the longer-term to hire highly skilled managerial, scientific and administrative personnel to fully implement our business plan and growth strategies. Due to the specialized scientific nature of our business, we are highly dependent upon our ability to attract and retain qualified scientific, technical and managerial personnel. Competition for these individuals, especially in San Diego, California, where many biotechnology companies are located, is intense and we may not be able to attract, assimilate or retain additional highly qualified personnel in the future. We may not be able to engage the services of qualified personnel at competitive prices or at all, particularly given the risks of employment attributable to our limited financial resources and lack of an established track record. Also, if we are required to attract personnel from other parts of the U.S. or abroad, we may have significant difficulty doing so due to the high cost of living in the Southern California area and due to the costs incurred with transferring personnel to the area. If we cannot attract and retain qualified staff and executives, we will be unable to develop our products and achieve regulatory clearance, and our business could fail.
We plan to expand our operations, which may strain our resources; our inability to manage our growth could delay or derail implementation of our business objectives.
We will need to significantly expand our operations to implement our longer-term business plan and growth strategies. We will also be required to manage multiple relationships with various strategic partners, technology licensors, customers, manufacturers and suppliers, consultants and other third parties. This expansion and these expanded relationships will require us to significantly improve or replace our existing managerial, operational and financial systems, procedures and controls; to improve the coordination between our various corporate functions; and to manage, train, motivate and maintain a growing employee base. The time and costs to effectuate these steps may place a significant strain on our management personnel, systems and resources, particularly given the limited amount of financial resources and skilled employees that may be available at the time. We may not be able to institute, in a timely manner or at all, the improvements to our managerial, operational and financial systems, procedures and controls necessary to support our anticipated increased levels of operations and to coordinate our various corporate functions, or that we may not be able to properly manage, train, motivate and retain our anticipated increased employee base. If we cannot manage our growth initiatives, including our expansion of our clinical trials in India and potentially in other countries, we will be unable to commercialize our products on a large-scale in a timely manner, if at all, and our business could fail.
| 21 |
We have limited experience in the organ transplant market and face competition from entities more familiar with this business and our efforts may not succeed.
We have investigated whether the Hemopurifier, when incorporated into a machine perfusion organ preservation circuit, can remove harmful viruses, exosomes, RNA molecules, cytokines, chemokines and other inflammatory molecules from recovered organs. This area is new to our product development and management personnel, and we may not be successful in the organ transplant market where we have limited experience. Even if we are successful in developing our Hemopurifier for the organ transplant market, we may not be able to compete effectively or generate significant revenues in this new area. Many companies of all sizes, including major pharmaceutical companies, specialized biotechnology companies, and traditional healthcare providers, are engaged in redesigning organ transplant care. Competitors operating in this area may have substantially greater financial and other resources, larger research and development staff, and more experience in this area. It is possible that, even if we are successful in the organ transplant field, that the market will not accept our product, or that our product will not generate significant revenues for us.
As a public company with limited financial resources undertaking the launch of new medical technologies, we may have difficulty attracting and retaining executive management and directors.
The directors and management of publicly traded corporations are increasingly concerned with the extent of their personal exposure to lawsuits and stockholder claims, as well as governmental and creditor claims which may be made against them, particularly in view of recent changes in securities laws imposing additional duties, obligations and liabilities on management and directors. Due to these perceived risks, directors and management are also becoming increasingly concerned with the availability of directors’ and officers’ liability insurance to pay on a timely basis the costs incurred in defending such claims. While we currently carry directors’ and officers’ liability insurance, such insurance is expensive and could be difficult to maintain in the future. If we are unable to continue or provide directors’ and officers’ liability insurance at affordable rates or at all, it may become increasingly more difficult to attract and retain qualified outside directors to serve on our Board of Directors. We may lose potential independent board members and management candidates to other companies in the biotechnology field that have greater directors’ and officers’ liability insurance to insure them from liability or to biotechnology companies that have revenues or have received greater funding to date which can offer greater compensation packages. The fees of directors are also rising in response to their increased duties, obligations and liabilities. In addition, our products could potentially be harmful to users, and we are exposed to claims of product liability including for injury or death. We have limited insurance and may not be able to afford robust coverage even as our products are introduced into the market. As a company with limited resources and potential exposures to management, we will have a more difficult time attracting and retaining management and outside independent directors than a more established public or private company due to these enhanced duties, obligations and potential liabilities.
If we fail to comply with extensive regulations of U.S. and foreign regulatory agencies, the commercialization of our products could be delayed or prevented entirely.
Our Hemopurifier product is subject to extensive government regulations related to development, testing, manufacturing and commercialization in the United States and other countries. The determination of when and whether a product is ready for large-scale purchase and potential use will be made by the U.S. Government through consultation with a number of governmental agencies, including the FDA, the National Institutes of Health, the CDC and the Department of Homeland Security. Our Hemopurifier has not received required regulatory approval from the FDA, or any foreign regulatory agencies, to be commercially marketed and sold. The process of obtaining and complying with FDA and other governmental regulatory approvals and regulations in the United States and in foreign countries is costly, time consuming, uncertain and subject to unanticipated delays. Obtaining such regulatory approvals, if any, can take several years. Despite the time and expense exerted, regulatory approval is never guaranteed. We also are subject to the following risks and obligations, among others:
| · | the FDA may refuse to approve an application if it believes that applicable regulatory criteria are not satisfied; | |
| · | the FDA may require additional testing for safety and effectiveness; | |
| · | the FDA may interpret data from pre-clinical testing and clinical trials in different ways than we interpret them; | |
| · | if regulatory approval of a product is granted, the approval may be limited to specific indications or limited with respect to its distribution; and | |
| · | the FDA may change its approval policies and/or adopt new regulations. |
| 22 |
Failure to comply with these or other regulatory requirements of the FDA may subject us to administrative or judicially imposed sanctions, including:
| · | warning letters; | |
| · | civil penalties; | |
| · | criminal penalties; | |
| · | injunctions; | |
| · | product seizure or detention; | |
| · | product recalls; and | |
| · | total or partial suspension of productions. |
Delays in successfully commencing or completing our planned clinical trials could jeopardize our ability to obtain regulatory approval and sustain our operations.
Our business prospects depend on our ability to complete studies, commence and complete our planned clinical trials, including our ongoing and planned studies in solid tumors in cancer, obtain satisfactory results, obtain required regulatory approvals and successfully commercialize our Hemopurifier product candidate. Completion of our clinical trials, announcement of results of the trials and our ability to obtain regulatory approvals could be delayed for a variety of reasons, including:
| · | failure to obtain required approvals to commence our planned clinical trials; | |
| · | slow patient enrollment in our planned clinical trials; | |
| · | serious adverse events related to our Hemopurifier; | |
| · | unsatisfactory results of any clinical trial; | |
| · | the failure of our principal third-party investigators to perform our clinical trials on our anticipated schedules; and | |
| · | different interpretations of our pre-clinical and clinical data, which could initially lead to inconclusive results. |
Our development costs will increase if we have material delays in any clinical trial or if we need to perform more or larger clinical trials than planned. If the delays are significant, or if any of our product candidates do not prove to be safe or effective or do not receive required regulatory approvals, our financial results and the commercial prospects for our product candidates will be harmed. Furthermore, our inability to complete our clinical trials in a timely manner could jeopardize our ability to obtain regulatory approval for our Hemopurifier or any other potential product candidates.
| 23 |
If we or our suppliers fail to comply with ongoing FDA or foreign regulatory authority requirements, or if we experience unanticipated problems with our products, these products could be subject to restrictions or withdrawal from the market.
Any product for which we obtain clearance or approval, if any, and the manufacturing processes, reporting requirements, post-approval clinical data and promotional activities for such product, will be subject to continued regulatory review, oversight and periodic inspections by the FDA and other domestic and foreign regulatory bodies. In particular, we and our third-party suppliers may be required to comply with the FDA’s Quality System Regulation, or QSR. These FDA regulations cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging, sterilization, storage and shipping of our products. Compliance with applicable regulatory requirements is subject to continual review and is monitored rigorously through periodic inspections by the FDA. If we, or our manufacturers, fail to adhere to QSR requirements in the United States, this could delay production of our products and lead to fines, difficulties in obtaining regulatory clearances, recalls, enforcement actions, including injunctive relief or consent decrees, or other consequences, which could, in turn, have a material adverse effect on our financial condition or results of operations.
In addition, the FDA assesses compliance with the QSR through periodic announced and unannounced inspections of manufacturing and other facilities. The failure by us or one of our suppliers to comply with applicable statutes and regulations administered by the FDA, or the failure to timely and adequately respond to any adverse inspectional observations or product safety issues, could result in any of the following enforcement actions:
| · | untitled letters, warning letters, fines, injunctions, consent decrees and civil penalties; | |
| · | unanticipated expenditures to address or defend such actions; | |
| · | customer notifications or repair, replacement, refunds, recall, detention or seizure of our products; | |
| · | operating restrictions or partial suspension or total shutdown of production; | |
| · | refusing or delaying our requests for 510(k) clearance or premarket approval of new products or modified products; | |
| · | withdrawing 510(k) clearances or premarket approvals that have already been granted; | |
| · | refusal to grant export approval for our products; or | |
| · | criminal prosecution. |
Moreover, the FDA strictly regulates the promotional claims that may be made about approved products. In particular, a product may not be promoted for uses that are not approved by the FDA as reflected in the product’s approved labeling. However, companies may share truthful and not misleading information that is otherwise consistent with a product’s FDA approved labeling. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant civil, criminal and administrative penalties.
Any of these sanctions could have a material adverse effect on our reputation, business, results of operations and financial condition. Furthermore, our key component suppliers may not currently be or may not continue to be in compliance with all applicable regulatory requirements, which could result in our failure to produce our products on a timely basis and in the required quantities, if at all.
| 24 |
If our products, or malfunction of our products, cause or contribute to a death or a serious injury, we will be subject to medical device reporting regulations, which can result in voluntary corrective actions or agency enforcement actions.
Under the FDA medical device reporting regulations, medical device manufacturers are required to report to the FDA information that a device has or may have caused or contributed to a death or serious injury or has malfunctioned in a way that would likely cause or contribute to death or serious injury if the malfunction of the device or one of our similar devices were to recur. If we fail to report these events to the FDA within the required timeframes, or at all, the FDA could take enforcement action against us. Any such adverse event involving our products also could result in future voluntary corrective actions, such as recalls or customer notifications, or agency action, such as inspection or enforcement action. Any corrective action, whether voluntary or involuntary, as well as defending ourselves in a lawsuit, will require the dedication of our time and capital, distract management from operating our business, and may harm our reputation and financial results.
We outsource many of our operational and development activities, and if any party to which we have outsourced certain essential functions fails to perform its obligations under agreements with us, the development and commercialization of our Hemopurifier product candidate and any future product candidates that we may develop could be delayed or terminated.
We rely on third-party consultants or other vendors to manage and implement much of the day-to-day conduct of our clinical trials and the manufacturing of our Hemopurifier product candidate. Accordingly, we are and will continue to be dependent on the timeliness and effectiveness of the efforts of these third parties. Our dependence on third parties includes key suppliers and third-party service providers supporting the development, manufacture and regulatory approval of our Hemopurifier, as well as support for our information technology systems and other infrastructure. While our management team oversees these vendors, failure of any of these third parties to meet their contractual, regulatory and other obligations or the development of factors that materially disrupt the performance of these third parties could have a material adverse effect on our business. For example, all of the key oversight responsibilities for the development and manufacture of our Hemopurifier are conducted by our management team, but all other activities are the responsibility of third-party vendors.
If a clinical research organization that we utilize is unable to allocate sufficient qualified personnel to our studies in a timely manner or if the work performed by it does not fully satisfy the requirements of the FDA or other regulatory agencies, we may encounter substantial delays and increased costs in completing our development efforts. Any manufacturer that we select may encounter difficulties in the manufacture of new products in commercial quantities, including problems involving product yields, product stability or shelf life, quality control, adequacy of control procedures and policies, compliance with FDA regulations and the need for further FDA approval of any new manufacturing processes and facilities. If any of these occur, the development and commercialization of our Hemopurifier product candidate could be delayed, curtailed or terminated, because we may not have sufficient financial resources or capabilities to continue such development and commercialization on our own.
If we or our contractors or service providers fail to comply with regulatory laws and regulations, we or they could be subject to regulatory actions, which could affect our ability to develop, market and sell our Hemopurifier product candidate and any other future product candidates that we may develop, if any, and may harm our reputation.
If we or our manufacturers or other third-party contractors fail to comply with applicable federal, state or foreign laws or regulations, we could be subject to regulatory actions, which could affect our ability to successfully develop, market and sell our Hemopurifier product candidate or any future product candidates, if any, and could harm our reputation and lead to reduced or non-acceptance of our proposed product candidates by the market. Even technical recommendations or evidence by the FDA through letters, site visits, and overall recommendations to academia or biotechnology companies may make the manufacturing of a clinical product extremely labor intensive or expensive, making the product candidate no longer viable to manufacture in a cost-efficient manner. The mode of administration may make the product candidate not commercially viable. The required testing of the product candidate may make that candidate no longer commercially viable. The conduct of clinical trials may be critiqued by the FDA, or a clinical trial site’s IRB or Institutional Biosafety Committee, which may delay or make impossible clinical testing of a product candidate. The IRB for a clinical trial may stop a trial or deem a product candidate unsafe to continue testing. This would have a material adverse effect on the value of the product candidate and our business prospects.
| 25 |
We will need to outsource and rely on third parties for the clinical development, sales and marketing of our Hemopurifier or any future product candidates that we may develop, and our future success will be dependent on the timeliness and effectiveness of the efforts of these third parties.
We do not have the required financial and human resources to carry out on our own all the pre-clinical and clinical development for our Hemopurifier product candidate or any other or future product candidates that we may develop, and do not have the capability and resources to market or sell our Hemopurifier product candidate or any future product candidates that we may develop. Our business model calls for the partial or full outsourcing of the clinical and other development, sales and marketing of our product candidates in order to reduce our capital and infrastructure costs as a means of potentially improving our financial position. Our success will depend on the performance of these outsourced providers. If these providers fail to perform adequately, our development of product candidates may be delayed and any delay in the development of our product candidates would have a material and adverse effect on our business prospects.
We are and will be exposed to product liability risks, and clinical and preclinical liability risks, which could place a substantial financial burden upon us should we be sued.
Our business exposes us to potential product liability and other liability risks that are inherent in the testing, manufacturing and marketing of medical devices. Claims may be asserted against us. A successful liability claim or series of claims brought against us could have a material adverse effect on our business, financial condition and results of operations. We may not be able to continue to obtain or maintain adequate product liability insurance on acceptable terms, if at all, and such insurance may not provide adequate coverage against potential liabilities. Claims or losses in excess of any product liability insurance coverage that we may obtain could have a material adverse effect on our business, financial condition and results of operations.
Our Hemopurifier product candidate may be used in connection with medical procedures in which it is important that those products function with precision and accuracy. If our product candidates, including our Hemopurifier, do not function as designed, or are designed improperly, we may be forced by regulatory agencies to withdraw such products from the market. In addition, if medical personnel or their patients suffer injury as a result of any failure of our products to function as designed, or our products are designed inappropriately, we may be subject to lawsuits seeking significant compensatory and punitive damages. The risk of product liability claims, product recalls and associated adverse publicity is inherent in the testing, manufacturing, marketing and sale of medical products. We have obtained general clinical trial liability insurance coverage. However, our insurance coverage may not be adequate or available. We may not be able to secure product liability insurance coverage on acceptable terms or at reasonable costs when needed. Any product recall or lawsuit seeking significant monetary damages may have a material effect on our business and financial condition. Any liability for mandatory damages could exceed the amount of our coverage. Moreover, a product recall could generate substantial negative publicity about our products and business and inhibit or prevent commercialization of other future product candidates.
We have not received, and may never receive, approval from the FDA to market a medical device in the United States.
Before a new medical device can be marketed in the United States, it must first receive a PMA or 510(k) clearance from the FDA, unless an exemption applies. A PMA submission, which is a higher standard than a 510(k) clearance, is used to demonstrate to the FDA that a new or modified device is safe and effective. The 510(k) is used to demonstrate that a device is “substantially equivalent” to a predicate device, that is, one that has been cleared by the FDA. We expect that any product we seek regulatory approval for, including the Hemopurifier, will require a PMA. The FDA approval process involves, among other things, successfully completing clinical trials and filing for and obtaining a PMA. The PMA process requires us to prove the safety and effectiveness of our products to the FDA’s satisfaction. This process, which includes preclinical studies and clinical trials, can take many years and requires the expenditure of substantial resources and may include post-marketing surveillance to establish the safety and efficacy of the product. Notwithstanding the effort and expense incurred, the process may never result in the FDA granting a PMA. Data obtained from preclinical studies and clinical trials are subject to varying interpretations that could delay, limit or prevent regulatory approval. Delays or rejections may also be encountered based upon changes in governmental policies for medical devices during the period of product development. The FDA can delay, limit or deny approval of a PMA application for many reasons, including:
| · | our inability to demonstrate safety or effectiveness of the Hemopurifier, or any other product we develop, to the FDA’s satisfaction; | |
| · | insufficient data from our preclinical studies and clinical trials, including for our Hemopurifier, to support approval; |
| 26 |
| · | failure of the facilities of our third-party manufacturer or suppliers to meet applicable requirements; | |
| · | inadequate compliance with preclinical, clinical or other regulations; | |
| · | our failure to meet the FDA’s statistical requirements for approval; and | |
| · | changes in the FDA’s approval policies, or the adoption of new regulations that require additional data or additional clinical trials. |
Modifications to products that are approved through a PMA application generally need FDA approval. Similarly, some modifications made to products cleared through a 510(k) may require a new 510(k). The FDA’s 510(k) clearance process usually takes from three to 12 months, but may last longer. The process of obtaining a PMA is much costlier and more uncertain than the 510(k) clearance process and generally takes from one to three years, or even longer, from the time the application is submitted to the FDA until an approval is obtained. Any of our products considered to be a class III device, which are considered to pose the greatest risk and the approval of which is governed by the strictest guidelines, will require the submission and approval of a PMA in order for us to market it in the United States. We also may design new products in the future that could require the clearance of a 510(k).
Although we have received approval to proceed with clinical trials of the Hemopurifier in the United States under the investigational device exemption, the current approval from the FDA to proceed could be revoked, the study could be unsuccessful, or the FDA PMA approval may not be obtained or could be revoked. Even if we obtain approval, the FDA or other regulatory authorities may require expensive or burdensome post-market testing or controls. Any delay in, or failure to receive or maintain, clearance or approval for our future products could prevent us from generating revenue from these products or achieving profitability. Additionally, the FDA and other regulatory authorities have broad enforcement powers. Regulatory enforcement or inquiries, or other increased scrutiny on us, could dissuade some physicians from using our products and adversely affect our reputation and the perceived safety and efficacy of our products.
The approval requirements for medical products used to fight bioterrorism and pandemics are still evolving, and any products we develop for such uses may not meet these requirements.
We are advancing product candidates under governmental policies that regulate the development and commercialization of medical treatment countermeasures against bioterror and pandemic threats. While we intend to pursue FDA market clearance to treat infectious bioterror and pandemic threats, it is often not feasible to conduct human studies against these deadly high threat pathogens. For example, the Hemopurifier is an investigational device that has not yet received FDA approval for any indication. We continue to investigate the potential for the use of the Hemopurifier in viral diseases under an open IDE and our FDA Breakthrough Designation for “…the treatment of life-threatening glycosylated viruses that are not addressed with an approved therapy.” We currently have an open FDA approved Expanded Access Protocol for the treatment of Ebola infected patients in the United States and a corresponding HealthCanada approval in Canada. Based on our studies to date, the Hemopurifier can potentially clear many viruses that are pathogenic in humans, including HCV, HIV, Monkeypox and Ebola.
For example, in June 2020, the FDA approved a supplement to our open IDE for the Hemopurifier in viral disease to allow for the testing of the Hemopurifier in patients with SARS-CoV-2/COVID-19 in a New Feasibility Study. This study was designed to enroll up to 40 subjects at up to 20 centers in the United States. Subjects had to have an established laboratory diagnosis of COVID-19, be admitted to an intensive care unit, or ICU, and have had acute lung injury and/or severe or life-threatening disease, among other criteria. Due to lack of COVID-19 patients in the ICUs of our trial sites, we terminated this study in 2022.
As a result of the termination of our COVID-19 study due to lack of patients in the ICUs, we were unable to demonstrate the effectiveness of our treatment countermeasures through controlled human efficacy studies in this U.S. study. Additionally, a change in government policies could impair our ability to obtain regulatory approval for the Hemopurifier.
| 27 |
The results of our clinical trials may not support our product candidate claims or may result in the discovery of adverse side effects.
Any research and development, pre-clinical testing and clinical trial activities involving our Hemopurifier and any additional products that we may develop are subject to extensive regulation and review by numerous governmental authorities both in the United States and abroad. Clinical studies must be conducted in compliance with FDA regulations or the FDA may take enforcement action. The data collected from these clinical studies may ultimately be used to support market clearance for these products. Even if our clinical trials are completed as planned, the results of these trials may not support our product candidate claims and the FDA may not agree with our conclusions regarding the trial results. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will be successful, and the later trials may not replicate the results of prior trials and pre-clinical studies. The clinical trial process may fail to demonstrate that our product candidates are safe and effective for the proposed indicated uses, which could cause us to abandon a product candidate and may delay development of others. Any delay or termination of our clinical trials will delay the filing of our product submissions and, ultimately, our ability to commercialize our product candidates and generate revenues. It is also possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of the product candidate’s profile.
U.S. legislative or FDA regulatory reforms may make it more difficult and costly for us to obtain regulatory approval of our product candidates and to manufacture, market and distribute our products after approval is obtained.
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the regulatory approval, manufacture and marketing of regulated products or the reimbursement thereof. In addition, FDA regulations and guidance are often revised or reinterpreted by the FDA in ways that may significantly affect our business and our products. Any new regulations or revisions or reinterpretations of existing regulations may impose additional costs or lengthen review times of future products. It is impossible to predict whether legislative changes will be enacted or FDA regulations, guidance or interpretations changed, and what the impact of such changes, if any, may be on our product development efforts.
Our current and future business activities are subject to applicable anti-kickback, fraud and abuse, false claims, physician payment transparency, health information privacy and security and other healthcare laws and regulations, which could expose us to significant penalties.
We are currently and will in the future be subject to healthcare regulation and enforcement by the U.S. federal government and the states in which we will conduct our business if our product candidates are approved by the FDA and commercialized in the United States. In addition to the FDA’s restrictions on marketing of approved products, the U.S. healthcare laws and regulations that may affect our ability to operate include: the federal fraud and abuse laws, including the federal anti-kickback and false claims laws; federal data privacy and security laws; and federal transparency laws related to payments and/or other transfers of value made to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors) and other healthcare professionals (such as physicians assistants and nurse practitioners) and teaching hospitals. Many states have similar laws and regulations that may differ from each other and federal law in significant ways, thus complicating compliance efforts. These laws may adversely affect our sales, marketing and other activities with respect to any product candidate for which we receive approval to market in the United States by imposing administrative and compliance burdens on us.
Because of the breadth of these laws and the narrowness of available statutory exceptions and regulatory safe harbors, it is possible that some of our business activities, particularly any sales and marketing activities after a product candidate has been approved for marketing in the United States, could be subject to legal challenge and enforcement actions. If our operations are found to be in violation of any of the federal and state laws described above or any other governmental regulations that apply to us, we may be subject to significant civil, criminal, and administrative penalties, including, without limitation, damages, fines, imprisonment, exclusion from participation in government healthcare programs, additional reporting obligations and oversight if we become subject to a corporate integrity agreement or other agreement to resolve allegations of non-compliance with these laws, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
| 28 |
We and the third parties with whom we work are subject to stringent and changing U.S. and foreign laws, rules, regulations and standards as well as policies, contracts and other obligations related to data privacy and security. Our actual or perceived failure to comply with such obligations, or such failure by the third parties with whom we work, could lead to regulatory investigations or actions, fines and penalties, a disruption of our clinical trials or commercialization of our products, private litigation, including class claims, and mass arbitration demands, harm to our reputation, or other adverse effects on our business or prospects.
In the ordinary course of business, we collect, receive, store, process, use, generate, transfer, disclose, make accessible, protect, secure, dispose of, transmit, and share, or collectively, “Process” or “Processing” personal data and other Sensitive Information (as defined below), including proprietary and confidential business data, trade secrets, and intellectual property that we collect in connection with clinical trials, as necessary to operate our business, for legal and marketing purposes, and for other business-related purposes. Our data Processing activities may subject us to numerous data privacy and security obligations, such as various laws, regulations, guidance, industry standards, external and internal privacy and security policies, representations, certifications, standards, publications, frameworks, contractual requirements and other obligations related to data privacy and security collectively, “Data Protection Obligations”.
In the United States, federal, state, and local governments have enacted numerous data privacy and security laws, including data breach notification laws, personal data privacy laws, consumer protection laws (e.g., Section 5 of the Federal Trade Commission Act), and other similar laws (e.g., wiretapping laws). For example, the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, imposes specific requirements relating to the privacy, security, and transmission of individually identifiable health information.
In addition, over the past few years, numerous U.S. states—including California, Virginia, Colorado, Connecticut, and Utah—have enacted comprehensive privacy laws that impose certain obligations on covered businesses, including providing specific disclosures in privacy notices and affording residents with certain rights concerning their personal data. As applicable, such rights may include the right to access, correct, or delete certain personal data, and to opt-out of certain data processing activities, such as targeted advertising, profiling, and automated decision-making. The exercise of these rights may impact our business and ability to provide our products and services. Certain states also impose stricter requirements for processing certain personal data, including sensitive information, such as conducting data privacy impact assessments. These state laws allow for statutory fines for noncompliance. For example, the California Consumer Privacy Act of 2018, or CCPA, applies to personal data of consumers, business representatives, and employees who are California residents, and requires covered businesses to provide specific disclosures in privacy notices and honor requests of California residents to exercise certain privacy rights. The CCPA also provides for fines of up to $7,500 per intentional violation and allows private litigants affected by certain data breaches to recover significant statutory damages. The CCPA and other comprehensive U.S. state privacy laws exempt some data Processing in the context of clinical trials, but these developments may further complicate compliance efforts, and increase legal risk and compliance costs for us, the third parties with whom we work. Similar laws are being considered in several other states, as well as at the federal and local levels, and we expect more states to pass similar laws in the future.
Outside the United States, an increasing number of laws, regulations, and industry standards may govern data privacy and security. For example, the European Union’s General Data Protection Regulation, or EU GDPR, and the United Kingdom’s GDPR, or UK GDPR, or collectively GDPR, Australia’s Privacy Act, and India’s Information Technology Act and supplementary rules impose strict requirements for Processing personal data. For example, under GDPR, companies can face private litigation related to Processing of personal data brought by classes of data subjects or consumer protection organizations authorized at law to represent their interests, temporary or definitive restrictions on data Processing or other corrective actions, and fines of up to the greater of 20 million Euros under the EU GDPR / 17.5 million pounds streamline under the UK GDPR or 4% of their worldwide annual revenue, whichever is greater.
| 29 |
In addition, we may be unable to transfer personal data from Europe and other jurisdictions to the United States or other countries due to data localization requirements or limitations on cross-border data flows. Europe and other jurisdictions have enacted laws requiring data to be localized or limiting the transfer of personal data to other countries. In particular, the European Economic Area, or EEA, and the United Kingdom, or UK, have significantly restricted the transfer of personal data to the United States and other countries whose privacy laws it generally believes are inadequate. Other jurisdictions may adopt similarly stringent interpretations of their data localization and cross-border data transfer laws. Although there are currently various mechanisms that may be used to transfer personal data from the EEA and UK to the United States in compliance with law, such as the EEA’s standard contractual clauses, the UK’s International Data Transfer Agreement / Addendum, and the EU-U.S. Data Privacy Framework and the UK extension thereto (which allows for transfers to relevant U.S.-based organizations who self-certify compliance and participate in the Framework) these mechanisms are subject to legal challenges, and there is no assurance that we can satisfy or rely on these measures to lawfully transfer personal data to the United States. If there is no lawful manner for us to transfer personal data from the EEA, the UK, or other jurisdictions to the United States, or if the requirements for a legally-compliant transfer are too onerous, we could face significant adverse consequences, including the interruption or degradation of our operations, the need to relocate part of or all of our business or data processing activities to other jurisdictions at significant expense, increased exposure to regulatory actions, substantial fines and penalties, the inability to transfer data and work with partners, vendors and other third parties, and injunctions against our processing or transferring of personal data necessary to operate our business. Some European regulators have ordered certain companies to suspend or permanently cease certain transfers of personal data out of Europe for allegedly violating the EU GDPR’s cross-border data transfer limitations. Additionally, companies that transfer personal data to recipients outside of the EEA and/or UK to other jurisdictions, particularly to the United States, are subject to increased scrutiny from regulators individual litigants and activist groups.
We publish privacy policies and may publish marketing materials and other statements, such as compliance with certain certifications or self-regulatory principles, regarding data privacy and security. If these policies, materials or statements are found to be deficient, lacking in transparency, deceptive, unfair, or misrepresentative of our practices, we may be subject to investigation, enforcement actions by regulators, or other adverse consequences.
In addition to data privacy and security laws, we are contractually subject to industry standards adopted by industry groups and may become subject to such obligations in the future. We are also bound by other contractual obligations related to data privacy and security, and our efforts to comply with such obligations may not be successful.
Data Protection Obligations, and consumers’ data privacy expectations, are quickly changing, becoming increasingly stringent, and creating uncertainty. Additionally, these obligations may be subject to differing applications and interpretations, which may be inconsistent or conflict among jurisdictions. Preparing for and complying with these obligations requires us to devote significant resources and may necessitate changes to our services, information technologies, systems, and practices and to those of any third parties that process personal data on our behalf.
Although we endeavor to comply with all applicable Data Protection Obligations, we may at times fail, or be perceived to have failed, to do so. Moreover, despite our efforts, our personnel or third parties with whom we work may fail to comply with such obligations, which could negatively impact our business operations and compliance posture.
If we or third parties fail, or are perceived to have failed, to address or comply with applicable Data Protection Obligations, it could: increase our compliance and operational costs; expose us to regulatory scrutiny, actions, fines and penalties; result in reputational harm; interrupt or stop our clinical trials; result in litigation and liability; result in an inability to process personal data or to operate in certain jurisdictions; harm our business operations or financial results or otherwise result in a material harm to our business, or other material adverse impact on our business, results of operations and financial condition. Additionally, given that Data Protection Obligations impose complex and burdensome obligations and that there is substantial uncertainty over the interpretation and application of these obligations, we may be required to incur material costs, divert management attention, and change our business operations, including our clinical trials, in an effort to comply, which could materially adversely affect our business operations and financial results.
Any of these events could have a material adverse effect on our reputation, business, or financial condition, including but not limited to: loss of customers; interruptions or stoppages in our business operations including, as relevant, clinical trials inability to process personal data or to operate in certain jurisdictions; limited ability to develop or commercialize our products; expenditure of time and resources to defend any claim or inquiry; adverse publicity; or revision or restructuring of our operations.
| 30 |
If our information technology systems, or those of third parties with whom we work, or our data are or were compromised, we could experience adverse consequences resulting from such compromise, including but not limited to: regulatory investigations or actions; litigation; fines and penalties; disruptions of our business operations; reputational harm; loss of revenue or profits; and other adverse consequences.
In the ordinary course of our business, we and third parties with whom we work may process proprietary, confidential and sensitive information, including personal data, intellectual property, trade secrets, and proprietary business information owned or controlled by ourselves or other third parties, or collectively, Sensitive Information. We may use and share Sensitive Information with service providers and subprocessors and other third parties with whom we work to help us operate our business. If we or such third parties with who we work have experienced, or in the future experience, any security incident(s) that result in any data loss; deletion or destruction; unauthorized access to; loss, unauthorized acquisition, disclosure, or exposure of, Sensitive Information, or other compromise related to the security, confidentiality, integrity of our, or their, information technology, software, services, communications or data, or collection, a Security Breach, it may result in an adverse impact on our business.
Cyberattacks, malicious internet-based activity and online and offline fraud are prevalent, continue to rise, and are increasingly difficult to detect. These threats come from a variety of sources, including traditional computer “hackers,” threat actors, “hacktivists,” organized criminal threat actors, personnel (such as through theft or misuse), sophisticated nation states, and nation-state-supported actors. Some actors now engage and are expected to continue to engage in cyber-attacks, including without limitation nation-state actors for geopolitical reasons and in conjunction with military conflicts and defense activities. During times of war and other major conflicts, we and the third parties with whom we work may be vulnerable to a heightened risk of these attacks, including retaliatory cyber-attacks, that could materially disrupt our systems and operations, supply chain, and ability to produce, sell and distribute our goods and services.
We and the third parties with whom we work are subject to a variety of evolving threats, including but not limited to social-engineering attacks, including through deep fakes, which may be increasingly more difficult to identify as fake, and phishing attacks, supply-chain attacks, loss of data or other information technology assets, adware, software bugs, malicious code, such as viruses and worms, employee theft or misuse, denial-of-service attacks, such as credential stuffing, and ransomware attacks. We may also be the subject of viruses, malware, including as a result of advanced persistent threat intrusions, server malfunction, software or hardware failures, loss of data or other computer assets, adware, attacks enhanced or facilitated by AI, telecommunications failures, earthquakes, fires, floods, or other similar threats.
Ransomware attacks, including by organized criminal threat actors, nation-states, and nation-state-supported actors, are becoming increasingly prevalent and severe, and can lead to significant interruptions in our operations, loss of Sensitive Information and income, reputational harm, and diversion of funds. Extortion payments may alleviate the negative impact of a ransomware attack, but we may be unwilling or unable to make such payments due to, for example, applicable laws or regulations prohibiting such payments.
Remote work has become more common and has increased risks to our information technology systems and data, as more of our employees utilize network connections, computers, and devices outside our premises or network, including working at home, while in transit and in public locations. Additionally, future or past business transactions, such as acquisitions or integrations, could expose us to additional cybersecurity risks and vulnerabilities, as our systems could be negatively affected by vulnerabilities present in acquired or integrated entities’ systems and technologies. Furthermore, we may discover security issues that were not found during due diligence of such acquired or integrated entities, and it may be difficult to integrate companies into our information technology environment and security program.
In addition, our reliance on third-party service providers could introduce new cybersecurity risks and vulnerabilities, including supply-chain attacks, and other threats to our business operations. We rely on third-parties and their technologies to operate critical business systems to process Sensitive Information in a variety of contexts, including, without limitation, cloud-based infrastructure, data center facilities, encryption and authentication technology, employee email, content delivery to customers, and other functions. We also rely on third-party service providers to assist with our clinical trials, provide other products or services, or otherwise to operate our business. Our ability to monitor these third parties’ information security practices is limited, and these third parties may not have adequate information security measures in place. If our third-party service providers experience a Security Breach or other interruption, we could experience adverse consequences. While we may be entitled to damages if our third-party service providers fail to satisfy their privacy or security-related obligations to us, any award may be insufficient to cover our damages, or we may be unable to recover such award. In addition, supply-chain attacks have increased in frequency and severity, and we cannot guarantee that third parties’ infrastructure in our supply chain or our third-party partners’ supply chains have not been compromised or that they do not contain exploitable defects or bugs that could result in a breach of or disruption to our information technology systems (including our services) or the third-party information technology systems that support us and our services.
| 31 |
While we have implemented security measures designed to protect against Security Breaches, these measures may not be effective. We take steps designed to detect, mitigate, and remediate vulnerabilities in our information technology systems, including our products, hardware and/or software, including that of third parties upon which we rely. We may not, however, detect or remediate all such vulnerabilities including on a timely basis. Further, we may experience delays in developing and deploying remedial measures and patched designed to address any such identified vulnerabilities. Vulnerabilities could be exploited and result in a security incident.
Any of the previously identified or similar threats could cause a Security Breach or other interruption and disrupt our ability and that of third parties with whom we work to provide our services.
We may expend significant resources, fundamentally change our business activities and practices, or modify our operations, including clinical trial activities, or information technology in an effort to protect against Security Breaches and to mitigate, detect and remediate actual and potential vulnerabilities. Applicable Data Protection Obligations may require us to implement specific security measures or use industry-standard or reasonable measures to protect against Security Breaches. Our security measures, or those of third parties with whom we work, may not be effective in protecting against Security Breaches.
Applicable Data Protection Obligations may require us to notify relevant stakeholders of Security Breaches, including affected individuals, customers, investors, partners, collaborators, regulators, law enforcement agencies and others, or to implement other requirements, such as providing credit monitoring. Such disclosures and compliance with such requirements are costly, and the disclosures or the failure to comply with such requirements could lead to an adverse impact on our business, results of operations and financial condition. If we or a third party with whom we work experiences a Security Breach or are perceived to have experienced a Security Breach, we may experience adverse consequences. These consequences may include: government enforcement actions, for example, investigations, fines, penalties, audits, and inspections; additional reporting requirements and/or oversight; restrictions on processing Sensitive Information, including personal data; litigation, including class claims; indemnification obligations; negative publicity; reputational harm; monetary fund diversions; diversion of management attention; interruptions in our operations, including availability of data; financial loss; and other similar harms. Security Breaches or other interruptions and attendant consequences may prevent or cause customers to stop using our services, deter new customers from using our services, and negatively impact our ability to grow and operate our business.
Our contracts may not contain limitations of liability, and even where they do, any such limitations or exclusions of liability in our contracts may not be adequate to protect us from liabilities or damages if we fail to comply with Data Protection Obligations related to information security or Security Breaches.
Our insurance coverage may not be adequate or otherwise protect us from or adequately mitigate liabilities or damages with respect to claims, costs, expenses, litigation, fines, penalties, business loss, data loss, regulatory actions or other material adverse impact on our business, results of operations and financial condition arising out of our Processing operations, privacy and security practices, or Security Breaches that we may experience. In addition, such coverage may not continue to be available on commercially reasonable terms or at all or be sufficient coverage to pay future claims. The successful assertion of one or more large claims against us that exceeds our available insurance coverage, or results in changes to our insurance policies, including premium increases or the imposition of large excess or deductible or co-insurance requirements, could have a material adverse impact on our business, results of operations and financial condition.
In addition to experiencing a Security Breach, third parties may gather, collect, or infer Sensitive Information about us from public sources, data brokers, or other means that reveals competitively sensitive details about our organization and could be used to undermine our competitive advantage or market position.
| 32 |
Should our products be approved for commercialization, lack of third-party coverage and reimbursement for our devices could delay or limit their adoption.
In both the U.S. and international markets, the use of medical devices is dependent in part on the availability of reimbursement from third-party payors, such as government and private insurance plans. Healthcare providers that use medical devices generally rely on third-party payors to pay for all or part of the costs and fees associated with the medical procedures being performed or to compensate them for their patient care services. Should our products under development be approved for commercialization by the FDA, any such products may not be considered cost-effective, reimbursement may not be available in the United States or other countries, if approved, and reimbursement may not be sufficient to allow sales of our future products, including the Hemopurifier, on a profitable basis. The coverage decisions of third-party payors will be significantly influenced by the assessment of our future products by health technology assessment bodies. These assessments are outside our control and any such evaluations may not be conducted or have a favorable outcome.
If approved for use in the United States, we expect that any products that we develop, including the Hemopurifier, will be purchased primarily by medical institutions, which will in turn bill various third-party payors for the health care services provided to patients at their facility. Payors may include the Centers for Medicare & Medicaid Services, or CMS, which administers the Medicare program and works in partnership with state governments to administer Medicaid, other government programs and private insurance plans. The process involved in applying for coverage and incremental reimbursement from CMS is lengthy and expensive. Further, Medicare coverage is based on our ability to demonstrate that the treatment is “reasonable and necessary” for Medicare beneficiaries. Even if products utilizing our Aethlon Hemopurifier technology receive FDA and other regulatory clearance or approval, they may not be granted coverage and reimbursement by any payor, including by CMS. For some governmental programs, such as Medicaid, coverage and adequate reimbursement differ from state to state and some state Medicaid programs may not pay adequate amounts for the procedure necessary to utilize products utilizing our technology system, or any payment at all. Moreover, many private payors use coverage decisions and payment amounts determined by CMS as guidelines in setting their coverage and reimbursement policies and amounts. However, no uniform policy requirement for coverage and reimbursement for medical devices exists among third-party payors in the United States. Therefore, coverage and reimbursement can differ significantly from payor to payor. If CMS or other agencies limit coverage or decrease or limit reimbursement payments for doctors and hospitals, this may affect coverage and reimbursement determinations by many private payors for any products that we develop.
Should our Hemopurifier or any future products, be approved for commercialization, certain health reform measures and adverse changes in reimbursement policies and procedures may impact our ability to market and sell our products.
Healthcare costs have risen significantly over the past decade, and there have been and continue to be proposals by legislators, regulators and third-party payors to decrease costs. Third-party payors are increasingly challenging the prices charged for medical products and services and instituting cost containment measures to control or significantly influence the purchase of medical products and services.
For example, in the United States, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act of 2010, or collectively, ACA, among other things, reduced and/or limited Medicare reimbursement to certain providers. On June 17, 2021, the U.S. Supreme Court dismissed a challenge on procedural grounds that argued the ACA is unconstitutional in its entirety because the “individual mandate” was repealed by Congress. Further, on August 16, 2022, President Biden signed the Inflation Reduction Act of 2022, or IRA, into law, which among other things, extends enhanced subsidies for individuals purchasing health insurance coverage in ACA marketplaces through plan year 2025. The IRA also eliminates the "donut hole" under the Medicare Part D program beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost and creating a new manufacturer discount program. It is unclear how any such challenges, and the healthcare reform measures of the Biden administration will impact the ACA and our business. The Budget Control Act of 2011, as amended by subsequent legislation, further reduces Medicare’s payments to providers by two percent through fiscal year 2032These reductions may reduce providers’ revenues or profits, which could affect their ability to purchase new technologies. Furthermore, the healthcare industry in the United States has experienced a trend toward cost containment as government and private insurers seek to control healthcare costs by imposing lower payment rates and negotiating reduced contract rates with service providers. In July 2021, the Biden Administration released an executive order, “Promoting Competition in the American Economy,” which contained provisions relating to prescription drugs. On September 9, 2021, in response to this executive order, the U.S. Department of Health and Human Services, or HHS, released a Comprehensive Plan for Addressing High Drug Prices that outlines principles for drug pricing reform and sets out a variety of potential legislative policies that Congress could pursue as well as potential administrative actions HHS can take to advance these principles. Further, the IRA, among other things (i) directs HHS to negotiate the price of certain high-expenditure, single-source drugs and biologics covered under Medicare and (ii) imposes rebates under Medicare Part B and Medicare Part D to penalize price increases that outpace inflation. These provisions will take effect progressively starting in fiscal year 2023, although they may be subject to legal challenges. HHS has and will continue to issue and update guidance as these programs are implemented. It is currently unclear how the IRA will be implemented but is likely to have a significant impact on the pharmaceutical industry. In addition, in response to the Biden administration’s October 2022 executive order, on February 14, 2023, HHS released a report outlining three new models for testing by the Center for Medicare and Medicaid Innovation which will be evaluated on their ability to lower the cost of drugs, promote accessibility, and improve quality of care. It is unclear whether the models will be utilized in any health reform measures in the future.
| 33 |
Legislation could be adopted in the future that limits payments for our products from governmental payors. In addition, commercial payors such as insurance companies, could adopt similar policies that limit reimbursement for medical device manufacturers’ products. Therefore, it is possible that our product or the procedures or patient care performed using our product will not be reimbursed at a cost-effective level. We face similar risks relating to adverse changes in reimbursement procedures and policies in other countries where we may market our products. Reimbursement and healthcare payment systems vary significantly among international markets. Our inability to obtain international reimbursement approval, or any adverse changes in the reimbursement policies of foreign payors, could negatively affect our ability to sell our products and have a material adverse effect on our business and financial condition.
Our ability to use net operating loss carryforwards and certain other tax attributes to offset future taxable income or taxes may be limited.
Under current law, federal net operating losses incurred in tax years beginning after December 31, 2017, may be carried forward indefinitely, but the deductibility of such federal net operating loss carryforwards in a taxable year is limited to 80% of taxable income in such year. In addition, under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, and corresponding provisions of state law, if a corporation undergoes an “ownership change,” which is generally defined as a greater than 50% change in its equity ownership value over a three-year period, the corporation’s ability to use its pre-change net operating loss carryforwards and other pre-change tax attributes to offset its post-change income or taxes may be limited. If we achieve profitability and an ownership change occurs and our ability to use our net operating loss carryforwards is materially limited, it would harm our future operating results by effectively increasing our future tax obligations. In addition, at the state level, there may be periods during which the use of net operating loss carryforwards is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed.
Uncertainties in the interpretation and application of existing, new and proposed tax laws and regulations could materially affect our tax obligations and effective tax rate.
The tax regimes to which we are subject or under which we operate are unsettled and may be subject to significant change. The issuance of additional guidance related to existing or future tax laws, or changes to tax laws or regulations proposed or implemented by the current or a future U.S. presidential administration, Congress, or taxing authorities in other jurisdictions, including jurisdictions outside of the United States, could materially affect our tax obligations and effective tax rate. To the extent that such changes have a negative impact on us, including as a result of related uncertainty, these changes may adversely impact our business, financial condition, results of operations, and cash flows.
The amount of taxes we pay in different jurisdictions depends on the application of the tax laws of various jurisdictions, including the United States, to our international business activities, tax rates, new or revised tax laws, or interpretations of tax laws and policies, and our ability to operate our business in a manner consistent with our corporate structure and intercompany arrangements. The taxing authorities of the jurisdictions in which we operate may challenge our methodologies for pricing intercompany transactions pursuant to our intercompany arrangements or disagree with our determinations as to the income and expenses attributable to specific jurisdictions. If such a challenge or disagreement were to occur, and our position was not sustained, we could be required to pay additional taxes, interest, and penalties, which could result in one-time tax charges, higher effective tax rates, reduced cash flows, and lower overall profitability of our operations. Our financial statements could fail to reflect adequate reserves to cover such a contingency. Similarly, a taxing authority could assert that we are subject to tax in a jurisdiction where we believe we have not established a taxable connection, often referred to as a “permanent establishment” under international tax treaties, and such an assertion, if successful, could increase our expected tax liability in one or more jurisdictions.
The Tax Cuts and Jobs Act of 2017 eliminated the option to deduct research and development expenses for tax purposes in the year incurred and requires taxpayers to capitalize and subsequently amortize such expenses over five years for research activities conducted in the United States and over 15 years for research activities conducted outside the United States. Although there have been legislative proposals to repeal or defer the capitalization requirement to later years, there can be no assurance that the provision will be repealed or otherwise modified. Future guidance from the Internal Revenue Service and other tax authorities with respect to such legislation may affect us, and certain aspects of such legislation could be repealed or modified in future legislation.
| 34 |
Our use of hazardous materials, chemicals and viruses exposes us to potential liabilities for which we may not have adequate insurance.
Our research and development involves the controlled use of hazardous materials, chemicals and viruses. The primary hazardous materials include chemicals needed to construct the Hemopurifier cartridges and the infected plasma samples used in preclinical testing of the Hemopurifier. All other chemicals are fully inventoried and reported to the appropriate authorities, such as the fire department, which inspects the facility on a regular basis. We are subject to federal, state, local and foreign laws governing the use, manufacture, storage, handling and disposal of such materials. Although we believe that our safety procedures for the use, manufacture, storage, handling and disposal of such materials comply with the standards prescribed by federal, state, local and foreign regulations, we cannot completely eliminate the risk of accidental contamination or injury from these materials. We have had no incidents or problems involving hazardous chemicals or biological samples. In the event of such an accident, we could be held liable for significant damages or fines.
We currently carry a limited amount of insurance to protect us from bodily injury or property damages arising from hazardous materials. Our product liability policy has a $5,000,000 limit of liability. For our facilities, our property policy provides $25,000 in coverage for contaminant clean-up or removal and $100,000 in coverage for damages to the premises resulting from contamination. Should we violate any regulations concerning the handling or use of hazardous materials, or should any injuries or death result from our use or handling of hazardous materials, we could be the subject of substantial lawsuits by governmental agencies or individuals. We may not have adequate insurance to cover all or any of such claims, if any. If we were responsible to pay significant damages for violations or injuries, if any, we might be forced to cease operations since such payments could deplete our available resources.
Our products may in the future be subject to product recalls. A recall of our products, either voluntarily or at the direction of the FDA or another governmental authority, including a third-country authority, or the discovery of serious safety issues with our products, could have a significant adverse impact on us.
The FDA and similar foreign governmental authorities have the authority to require the recall of commercialized products in the event of material deficiencies or defects in design or manufacture. For the FDA, the authority to require a recall must be based on a finding that there is reasonable probability that the device would cause serious injury or death. In addition, foreign governmental bodies have the authority to require the recall of our products in the event of material deficiencies or defects in design or manufacture. Manufacturers may, under their own initiative, recall a product if any material deficiency in a device is found. The FDA requires that certain classifications of recalls be reported to the FDA within ten working days after the recall is initiated. A government-mandated or voluntary recall by us or one of our international distributors could occur as a result of an unacceptable risk to health, component failures, malfunctions, manufacturing errors, design or labeling defects or other deficiencies and issues. Recalls of any of our products would divert managerial and financial resources and have an adverse effect on our reputation, results of operations and financial condition, which could impair our ability to produce our products in a cost-effective and timely manner in order to meet our customers’ demands. We may also be subject to liability claims, be required to bear other costs, or take other actions that may have a negative impact on our future sales and our ability to generate profits. Companies are required to maintain certain records of recalls, even if they are not reportable to the FDA or another third-country competent authority. We may initiate voluntary recalls involving our products in the future that we determine do not require notification of the FDA or another third-country competent authority. If the FDA disagrees with our determinations, they could require us to report those actions as recalls. A future recall announcement could harm our reputation with customers and negatively affect our sales. In addition, the FDA could take enforcement action for failing to report recalls. We are also required to follow detailed recordkeeping requirements for all firm-initiated medical device corrections and removals.
Even though we have received breakthrough device designation for the Hemopurifier for two independent indications, this designation may not expedite the development or review of the Hemopurifier and does not provide assurance ultimately of PMA submission or approval by the FDA.
The Breakthrough Devices Program is a voluntary program intended to expedite the review, development, assessment and review of certain medical devices that provide for more effective treatment or diagnosis of life-threatening or irreversibly debilitating human diseases or conditions for which no approved or cleared treatment exists or that offer significant advantages over existing approved or cleared alternatives. All submissions for devices designated as Breakthrough Devices will receive priority review, meaning that the review of the submission is placed at the top of the appropriate review queue and receives additional review resources, as needed.
| 35 |
Although breakthrough designation or access to any other expedited program may expedite the development or approval process, it does not change the standards for approval. Although we obtained breakthrough device designation for the Hemopurifier for two indications, we may not experience faster development timelines or achieve faster review or approval compared to conventional FDA procedures. For example, the time required to identify and resolve issues relating to manufacturing and controls, the acquisition of a sufficient supply of our product for clinical trial purposes or the need to conduct additional nonclinical or clinical studies may delay approval by the FDA, even if the product qualifies for breakthrough designation or access to any other expedited program. Access to an expedited program may also be withdrawn by the FDA if it believes that the designation is no longer supported by data from our clinical development program. Additionally, qualification for any expedited review procedure does not ensure that we will ultimately obtain regulatory approval for the product.
Our bylaws designate the Eighth Judicial District Court of Clark County, Nevada, as the sole and exclusive forum for certain types of actions and proceedings that may be initiated by our stockholders, which could limit our stockholders’ ability to obtain a favorable judicial forum for disputes with us or our directors, officers, employees or agents.
Our bylaws require that, to the fullest extent permitted by law, and unless the Company consents in writing to the selection of an alternative forum, the Eighth Judicial District Court of Clark County, Nevada, will, to the fullest extent permitted by law, be the sole and exclusive forum for each of the following:
| · | any derivative action or proceeding brought in the name or right of the Company or on its behalf, | |
| · | any action asserting a claim for breach of any fiduciary duty owed by any director, officer, employee or agent of the Company to the Company or the Company’s stockholders, | |
| · | any action arising or asserting a claim arising pursuant to any provision of NRS Chapters 78 or 92A or any provision of our articles of incorporation or bylaws, or | |
| · | any action asserting a claim governed by the internal affairs doctrine, including, without limitation, any action to interpret, apply, enforce or determine the validity of our articles of incorporation or bylaws. |
However, our bylaws provide that the exclusive forum provisions do not apply to suits brought to enforce any liability or duty created by the Exchange Act or any other claim for which the federal courts have exclusive jurisdiction. We note that there is uncertainty as to whether a court would enforce the provision and that investors cannot waive compliance with the federal securities laws and the rules and regulations thereunder. Although we believe this provision benefits us by providing increased consistency in the application of Nevada law in the types of lawsuits to which it applies, the provision may have the effect of discouraging lawsuits against our directors and officers.
Risks Related to Our Intellectual Property and Related Litigation
We rely upon licenses and patent rights from third parties which are subject to termination or expiration.
We rely in part upon third-party licenses and ownership rights assigned from third parties for the development of specific uses for our Hemopurifier devices. We are researching, developing and testing cancer-related applications for our devices under patents assigned from the London Health Science Center Research, Inc. Under the assignment agreement, we own the patents outright for the life of the patent, which expires in May 2029. Under certain circumstances, ownership of the patents may revert to the London Health Science Center Research, Inc. if there is an uncured substantial breach of the assignment agreement. Should any of our licenses be prematurely terminated for any reason, or if the patents and intellectual property assigned to us or owned by such entities that we have licensed are challenged or defeated by third parties, our research efforts could be materially and adversely affected. Our licenses and patents assigned to us may not continue in force for as long as we require for our research, development and testing of cancer treatments. It is possible that, if our licenses terminate or the underlying patents and intellectual property is challenged or defeated or the patents and intellectual property assigned to us is challenged or defeated, suitable replacements may not be obtained or developed on terms acceptable to us, if at all. There is also the related risk that we may not be able to make the required payments under any patent license or assignment agreement, in which case we may lose ability to use one or more of the licensed or assigned patents.
| 36 |
We could become subject to intellectual property litigation that could be costly, result in the diversion of management’s time and efforts, require us to pay damages, prevent us from selling our commercially available products and/or reduce the margins we may realize from our products.
The medical devices industry is characterized by extensive litigation and administrative proceedings over patent and other intellectual property rights. Whether a product infringes a patent involves complex legal and factual issues, and the determination is often uncertain. There may be existing patents of which we are unaware that our products under development may inadvertently infringe. The likelihood that patent infringement claims may be brought against us increases as the number of participants in the infectious market increases and as we achieve more visibility in the marketplace and introduce products to market.
Any infringement claim against us, even if without merit, may cause us to incur substantial costs, and would place a significant strain on our financial resources, divert the attention of management from our core business, and harm our reputation. In some cases, litigation may be threatened or brought by a patent holding company or other adverse patent owner who has no relevant product revenues and against whom our patents may provide little or no deterrence. If we are found to infringe any patents, we could be required to pay substantial damages, including triple damages if an infringement is found to be willful. We also could be required to pay royalties and could be prevented from selling our products unless we obtain a license or are able to redesign our products to avoid infringement. We may not be able to obtain a license enabling us to sell our products on reasonable terms, or at all. If we fail to obtain any required licenses or make any necessary changes to our technologies or the products, we may be unable to commercialize one or more of our products or may have to withdraw products from the market, all of which would have a material adverse effect on our business, financial condition and results of operations.
If the combination of patents, trade secrets and contractual provisions upon which we rely to protect our intellectual property is inadequate, our ability to commercialize our products successfully will be harmed.
Our success depends significantly on our ability to protect our proprietary rights to the technologies incorporated in our products. We currently have three issued U.S. patents and two pending U.S. patent applications. We also have 14 issued foreign patents and have applied for 15 additional foreign and international patents. Our issued patents begin to expire in March 2027, with the last of these patents expiring in 2031, although terminal disclaimers, patent term extension or patent term adjustment can shorten or lengthen the patent term. We rely on a combination of patent protection, trade secret laws and nondisclosure, confidentiality and other contractual restrictions to protect our proprietary technology. However, these may not adequately protect our rights or permit us to gain or keep any competitive advantage.
The issuance of a patent is not conclusive as to its scope, validity or enforceability. The scope, validity or enforceability of our issued patents can be challenged in litigation or proceedings before the U.S. Patent and Trademark Office or foreign patent offices where our applications are pending. The U.S. Patent and Trademark Office or foreign offices may deny or require significant narrowing of claims in our pending patent applications. Patents issued as a result of the pending patent applications, if any, may not provide us with significant commercial protection or be issued in a form that is advantageous to us. Proceedings before the U.S. Patent and Trademark Office or foreign offices could result in adverse decisions as to the priority of our inventions and the narrowing or invalidation of claims in issued patents. The laws of some foreign countries may not protect our intellectual property rights to the same extent as the laws of the U.S., if at all. Some of our patents may expire before we receive FDA approval to market our products in the United States or we receive approval to market our products in a foreign country. Although we believe that certain patent applications and/or other patents issued more recently will help protect the proprietary nature of the Hemopurifier treatment technology, this protection may not be sufficient to protect us during the development of that technology.
Our competitors may successfully challenge and invalidate or render unenforceable our issued patents, including any patents that may issue in the future, which could prevent or limit our ability to market our products and could limit our ability to stop competitors from marketing products that are substantially equivalent to ours. In addition, competitors may be able to design around our patents or develop products that provide outcomes that are comparable to our products but that are not covered by our patents.
We have also entered into confidentiality and assignment of intellectual property agreements with all of our employees, consultants and advisors directly involved in the development of our technology as one of the ways we seek to protect our intellectual property and other proprietary technology. However, these agreements may not be enforceable or may not provide meaningful protection for our trade secrets or other proprietary information in the event of unauthorized use or disclosure or other breaches of the agreements.
| 37 |
In the event a competitor infringes upon any of our patents or other intellectual property rights, enforcing our rights may be difficult, time consuming and expensive, and would divert management’s attention from managing our business. We may not be successful on the merits in any enforcement effort. In addition, we may not have sufficient resources to litigate, enforce or defend our intellectual property rights.
We may rely on licenses for new technology, which may affect our continued operations with respect thereto.
As we develop our technology, we may need to license additional technologies to optimize the performance of our products. We may not be able to license these technologies on commercially reasonable terms or at all. In addition, we may fail to successfully integrate any licensed technology into our proposed products. Our inability to obtain any necessary licenses could delay our product development and testing until alternative technologies can be identified, licensed and integrated. The inability to obtain any necessary third-party licenses could cause us to abandon a particular development path, which could seriously harm our business, financial position and results of our operations.
New technology may lead to our competitors developing superior products which would reduce demand for our products.
Research into technologies similar to ours is proceeding at a rapid pace, and many private and public companies and research institutions are actively engaged in the development of products similar to ours. These new technologies may, if successfully developed, offer significant performance or price advantages when compared with our technologies. Our existing patents or our pending and proposed patent applications may not offer meaningful protection if a competitor develops a novel product based on a new technology.
If we are unable to protect our proprietary technology and preserve our trade secrets, we will increase our vulnerability to competitors which could materially adversely impact our ability to remain in business.
Our ability to successfully commercialize our products will depend on our ability to protect those products and our technology with domestic and foreign patents. We will also need to continue to preserve our trade secrets. The issuance of a patent is not conclusive as to its validity or as to the enforceable scope of the claims of the patent. The patent positions of technology companies, including us, are uncertain and involve complex legal and factual issues. Our patents may not prevent other companies from developing similar products or products which produce benefits substantially the same as our products, and other companies may be issued patents that may prevent the sale of our products or require us to pay significant licensing fees in order to market our products.
From time to time, we may need to obtain licenses to patents and other proprietary rights held by third parties in order to develop, manufacture and market our products. If we are unable to timely obtain these licenses on commercially reasonable terms, our ability to commercially exploit such products may be inhibited or prevented. Our pending patent applications may not result in issued patents, patent protection may not be secured for any particular technology, and our issued patents may not be valid or enforceable or provide us with meaningful protection.
If we are required to engage in expensive and lengthy litigation to enforce our intellectual property rights, such litigation could be very costly and the results of such litigation may not be satisfactory.
Although we have entered into invention assignment agreements with our employees and with certain advisors, and we routinely enter into confidentiality agreements with our contract partners, if those employees, advisors or contract partners develop inventions or processes independently that may relate to products or technology under development by us, disputes may arise about the ownership of those inventions or processes. Time-consuming and costly litigation could be necessary to enforce and determine the scope of our rights under these agreements. In addition, we may be required to commence litigation to enforce such agreements if they are violated, and it is certainly possible that we will not have adequate remedies for breaches of our confidentiality agreements as monetary damages may not be sufficient to compensate us. We may be unable to fund the costs of any such litigation to a satisfactory conclusion, which could leave us without recourse to enforce contracts that protect our intellectual property rights.
| 38 |
Other companies may claim that our technology infringes on their intellectual property or proprietary rights and commence legal proceedings against us which could be time-consuming and expensive and could result in our being prohibited from developing, marketing, selling or distributing our products.
Because of the complex and difficult legal and factual questions that relate to patent positions in our industry, it is possible that our products or technology could be found to infringe upon the intellectual property or proprietary rights of others. Third parties may claim that our products or technology infringe on their patents, copyrights, trademarks or other proprietary rights and demand that we cease development or marketing of those products or technology or pay license fees. We may not be able to avoid costly patent infringement litigation, which will divert the attention of management away from the development of new products and the operation of our business. We may not prevail in any such litigation. If we are found to have infringed on a third-party’s intellectual property rights, we may be liable for money damages, encounter significant delays in bringing products to market or be precluded from manufacturing particular products or using particular technology.
Other parties may challenge certain of our foreign patent applications. If any such parties are successful in opposing our foreign patent applications, we may not gain the protection afforded by those patent applications in particular jurisdictions and may face additional proceedings with respect to similar patents in other jurisdictions, as well as related patents. The loss of patent protection in one jurisdiction may influence our ability to maintain patent protection for the same technology in other jurisdictions.
Risks Related to U.S. Government Contracts
We may not obtain U.S. Government contracts to further develop our technology.
While we have previously had U.S. government contracts, we may not be successful in obtaining future government grants or contracts. The process of obtaining government contracts is lengthy with the uncertainty that we will be successful in obtaining announced grants or contracts for therapeutics as a medical device technology. Accordingly, although we have obtained government contracts in the past, we may not be awarded any future U.S. Government grants or contracts utilizing our Hemopurifier platform technology.
U.S. Government agencies have special contracting requirements, including a right to audit us, which create additional risks; a negative audit would be detrimental to us.
Our business plan to utilize the Aethlon Hemopurifier technology may seek to involve contracts with the U.S. Government. Many government contracts, typically contain unfavorable termination provisions and are subject to audit and modification by the government at its sole discretion, which would subject us to additional risks should we obtain contracts with the U.S. Government in the future. These risks include the ability of the U.S. Government to unilaterally:
| · | suspend or prevent us for a period of time from receiving new contracts or extending existing contracts based on violations or suspected violations of laws or regulations; | |
| · | audit and object to our contract-related costs and fees, including allocated indirect costs; | |
| · | control and potentially prohibit the export of our products; and | |
| · | change certain terms and conditions in our contracts. |
| 39 |
As a former and potential future U.S. Government contractor, we are required to comply with applicable laws, regulations and standards relating to our accounting practices and would be subject to periodic audits and reviews. As part of any such audit or review, the U.S. Government may review the adequacy of, and our compliance with, our internal control systems and policies, including those relating to our purchasing, property, estimating, compensation and management information systems. Based on the results of its audits, the U.S. Government may adjust our contract-related costs and fees, including allocated indirect costs. In addition, if an audit or review uncovers any improper or illegal activity, we would possibly be subject to civil and criminal penalties and administrative sanctions, including termination of our contracts, forfeiture of profits, suspension of payments, fines and suspension or prohibition from doing business with the U.S. Government. We could also suffer serious harm to our reputation if allegations of impropriety were made against us. Although we have not had any government audits and reviews to date, future audits and reviews could cause adverse effects. In addition, under U.S. Government purchasing regulations, some of our costs, including most financing costs, amortization of intangible assets, portions of our research and development costs, and some marketing expenses, would possibly not be reimbursable or allowed under such contracts. Further, as a former and potential future U.S. Government contractor, we would be subject to an increased risk of investigations, criminal prosecution, civil fraud, whistleblower lawsuits and other legal actions and liabilities. Moreover, recent actions by the U.S. Congress and executive agencies to reduce discretionary spending, impose funding constraints, or shift policy priorities could limit the availability of government funding for programs that might otherwise support the development or acquisition of our technology. These developments could reduce the likelihood of future contract opportunities or result in the modification, delay, or cancellation of existing or anticipated government solicitations involving our products.
As a potential future U.S. Government contractor, we would be subject to a number of procurement rules and regulations.
Government contractors must comply with specific procurement regulations and other requirements. These requirements, although customary in government contracts, would impact our performance and compliance costs. In addition, current U.S. Government budgetary constraints could lead to changes in the procurement environment, including the Department of Defense’s initiative focused on efficiencies, affordability and cost growth and other changes to its procurement practices. If and to the extent such changes occur, they could affect whether and, if so, how we pursue certain opportunities and the terms under which we are able to do so.
In addition, failure to comply with these regulations and requirements could result in reductions of the value of contracts, contract modifications or termination, and the assessment of penalties and fines, which could negatively impact our results of operations and financial condition. Our failure to comply with these regulations and requirements could also lead to suspension or debarment, for cause, from government contracting or subcontracting for a period of time. Among the causes for debarment are violations of various statutes, including those related to procurement integrity, export control, government security regulations, employment practices, protection of the environment, accuracy of records and the recording of costs, and foreign corruption. The termination of any government contract we may obtain as a result of any of these acts could have a negative impact on our results of operations and financial condition and could have a negative impact on our reputation and ability to procure other government contracts in the future.
Risks Relating to Our Common Stock and Our Corporate Governance
If we are unable to maintain compliance with the listing requirements of the Nasdaq Capital Market, our common stock may be delisted from the Nasdaq Capital Market, which could have a material adverse effect on our financial condition and could make it more difficult for you to sell your shares.
Our common stock is listed on the Nasdaq Capital Market and we are therefore subject to its continued listing requirements, including requirements with respect to the market value of publicly held shares, market value of listed shares, minimum bid price per share (subject to a 180-day grace period, as discussed below) and minimum stockholders’ equity, among others, and requirements relating to board and committee independence. If we fail to satisfy one or more of the requirements, we may be delisted from the Nasdaq Capital Market.
On June 27, 2024, we received a letter, or Notice, from The Nasdaq Stock Market, or Nasdaq, that we were not in compliance with the $1.00 minimum bid price requirement for continued listing on the Nasdaq Capital Market, as set forth in Nasdaq Listing Rule 5550(a)(2), or the Minimum Bid Price Requirement. The Notice indicated that, consistent with Nasdaq Listing Rule 5810(c)(3)(A), we have 180 calendar days to regain compliance with the Minimum Bid Price Requirement by having the closing bid price of our common stock meet or exceed $1.00 per share for at least ten consecutive business days.
| 40 |
On January 7, 2025, the Company received a letter from Nasdaq (the “Extension Notice”) advising that the Company has been granted a 180-day extension, or until June 23, 2025, to regain compliance with the Minimum Bid Price Requirement, in accordance with Nasdaq Listing Rule 5810(c)(3)(A). If at any time prior to June 23, 2025, the bid price of the Company’s common stock closes at $1.00 per share or more for a minimum of 10 consecutive trading days, the Company will regain compliance with the Minimum Bid Price Requirement. On January 7, 2025, the Company received a letter from Nasdaq (the “Extension Notice”) advising that the Company has been granted a 180-day extension, or until June 23, 2025, to regain compliance with the Minimum Bid Price Requirement, in accordance with Nasdaq Listing Rule 5810(c)(3)(A). If at any time prior to June 23, 2025, the bid price of the Company’s common stock closes at $1.00 per share or more for a minimum of 10 consecutive trading days, the Company will regain compliance with the Minimum Bid Price Requirement.
Effective as of the close of business on June 6, 2025 with an effective trading date of June 9, 2025 the Company effectuated a 1-for-8 reverse split to attempt to maintain compliance with the $1.00 minimum bid requirement and to avoid, or at least mitigate, the likely adverse consequences of our common stock being delisted from the Nasdaq Capital Market by producing the immediate effect of increasing the bid price of our common stock.
On June 24, 2025, the Company received notification that they had regained compliance with the Minimum Bid Price Requirement.
There can be no assurance, however, that we will be able to continue to maintain compliance with the continued listing requirements for the Nasdaq Capital Market and our common stock could be delisted in the future. In addition, we may be unable to meet other applicable listing requirements of the Nasdaq Capital Market, including maintaining minimum levels of stockholders’ equity or market values of our common stock in which case, our common stock could be delisted notwithstanding our ability to demonstrate compliance with the Minimum Bid Price Requirement.
Delisting from the Nasdaq Capital Market may adversely affect our ability to raise additional financing through the public or private sale of equity securities, may significantly affect the ability of investors to trade our securities and may negatively affect the value and liquidity of our common stock. Delisting also could have other negative results, including the potential loss of employee confidence, the loss of institutional investors or interest in business development opportunities.
Historically we have not paid dividends on our common stock, and we do not anticipate paying any cash dividends in the foreseeable future.
We have never paid cash dividends on our common stock. We intend to retain our future earnings, if any, to fund operational and capital expenditure needs of our business, and do not anticipate paying any cash dividends in the foreseeable future. As a result, capital appreciation, if any, of our common stock will be the sole source of gain for our common stockholders in the foreseeable future.
Our stock price is speculative, and there is a risk of litigation.
The trading price of our common stock has in the past and may in the future be subject to wide fluctuations in response to factors such as the following:
| · | failure to raise additional funds when needed; | |
| · | announcements regarding our ongoing development of the Hemopurifier; | |
| · | results regarding the progress of our clinical trials with the Hemopurifier; | |
| · | results reported from our clinical trials with the Hemopurifier; | |
| · | failure to meet the continued listing requirements of and maintain our listing on Nasdaq; | |
| · | results of operations or revenue in any quarter failing to meet the expectations, published or otherwise, of the investment community; |
| 41 |
| · | reduced investor confidence in equity markets; | |
| · | speculation in the press or analyst community; | |
| · | wide fluctuations in stock prices, particularly with respect to the stock prices for other medical device companies; | |
| · | announcements of technological innovations by us or our competitors; | |
| · | new products or the acquisition of significant customers by us or our competitors; | |
| · | changes in interest rates; | |
| · | changes in investors’ beliefs as to the appropriate price-earnings ratios for us and our competitors; | |
| · | changes in recommendations or financial estimates by securities analysts who track our common stock or the stock of other medical device companies; | |
| · | changes in management; | |
| · | sales of common stock by directors and executive officers; | |
| · | rumors or dissemination of false or misleading information, particularly through Internet chat rooms, instant messaging, and other rapid-dissemination methods; | |
| · | conditions and trends in the medical device industry generally; | |
| · | the announcement of acquisitions or other significant transactions by us or our competitors; | |
| · | adoption of new accounting standards affecting our industry; | |
| · | changes in the structure of healthcare payment systems; | |
| · | general market conditions; | |
| social and geopolitical incidents and issues; | ||
| · | domestic or international terrorism and other factors; and | |
| · | the other factors described in this section. |
Fluctuations in the price of our common stock may expose us to the risk of securities class action lawsuits. Although no such lawsuits are currently pending against us and we are not aware that any such lawsuit is threatened to be filed in the future, future lawsuits are possible as a result of fluctuations in the price of our common stock. Defending against any such suits could result in substantial cost and divert management’s attention and resources. In addition, any settlement or adverse determination of such lawsuits could subject us to significant liability.
| 42 |
If at any time our common stock is subject to the SEC’s penny stock rules, broker-dealers may experience difficulty in completing customer transactions and trading activity in our securities may be adversely affected.
If at any time our common stock is not listed on a national securities exchange or we have net tangible assets of $2,000,000 or less, or we have an average revenue of less than $6,000,000 for the last three years, and our common stock has a market price per share of less than $5.00, transactions in our common stock will be subject to the SEC’s “penny stock” rules. Currently, our common stock is subject to the SEC’s “penny stock” rules promulgated under the Exchange Act and as a result, broker-dealers may find it difficult to effectuate customer transactions and trading activity in our securities may be adversely affected. For any transaction involving a penny stock, unless exempt, the rules require:
| · | that a broker or dealer approve a person’s account for transactions in penny stocks; | |
| · | furnish the investor a disclosure document describing the risks of investing in penny stocks; | |
| · | disclose to the investor the current market quotation, if any, for the penny stock; | |
| · | disclose to the investor the amount of compensation the firm and its broker will receive for the trade; and | |
| · | The broker or dealer receive from the investor a written agreement to the transaction, setting forth the identity and quantity of the penny stock to be purchased. |
In order to approve a person’s account for transactions in penny stocks, the broker or dealer must:
| · | obtain financial information and investment experience objectives of the person; and | |
| · | make a reasonable determination that the transactions in penny stocks are suitable for that person and the person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks. |
The broker or dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prescribed by the SEC relating to the penny stock market, which, in highlight form:
| · | sets forth the basis on which the broker or dealer made the suitability determination; and | |
| · | that the broker or dealer received a signed, written agreement from the investor prior to the transaction. |
Generally, brokers may be less willing to execute transactions in securities subject to the “penny stock” rules. This may make it more difficult for investors to dispose of our common stock and cause a decline in the market value of our stock.
Disclosure also has to be made about the risks of investing in penny stocks in both public offerings and in secondary trading and about the commissions payable to both the broker-dealer and the registered representative, current quotations for the securities and the rights and remedies available to an investor in cases of fraud in penny stock transactions. Finally, monthly statements have to be sent disclosing recent price information for the penny stock held in the account and information on the limited market in penny stocks.
| 43 |
Our common stock has had an unpredictable trading volume which means you may not be able to sell our shares at or near trading prices or at all.
Trading in our common shares historically has been volatile and often has been thin, meaning that the number of persons interested in purchasing our common shares at or near trading prices at any given time may be relatively small or non-existent. This situation is attributable to a number of factors, including the fact that we are a small company which is relatively unknown to stock analysts, stock brokers, institutional investors and others in the investment community that generate or influence sales volume, and that even if we came to the attention of such persons, they tend to be risk-averse and would be reluctant to follow an unproven company such as ours or purchase or recommend the purchase of our shares until such time as we became more seasoned and viable. As a consequence, there may be periods of several days or more when trading activity in our shares is minimal, as compared to a seasoned issuer which has a large and steady volume of trading activity that will generally support continuous sales without an adverse effect on share price. A broader or more active public trading market for our common shares may not develop or be sustained, and current trading levels may decrease.
The market price for our common stock is volatile; you may not be able to sell our common stock at or above the price you have paid for it, which may result in losses to you.
The market for our common stock is characterized by significant price volatility when compared to seasoned issuers, and we expect that our share price will continue to be more volatile than a seasoned issuer for the indefinite future. During the 52-week period ended March 31, 2025, the high and low closing sale prices for a share of our common stock were $14.08 and $2.08, respectively. The volatility in our share price is attributable to a number of factors. First, as noted above, trading in our common stock often has been thin. As a consequence of this lack of liquidity, the trading of relatively small quantities of shares by our stockholders may disproportionately influence the price of those shares in either direction. The price for our shares could, for example, decline precipitously in the event that a large number of our common shares are sold on the market without commensurate demand, as compared to a seasoned issuer which could better absorb those sales without adverse impact on its share price. Secondly, we are a speculative investment due to our limited operating history, limited amount of cash and revenue, lack of profit to date, and the uncertainty of future market acceptance for our potential products. As a consequence of this enhanced risk, more risk-adverse investors may, under the fear of losing all or most of their investment in the event of negative news or lack of progress, be more inclined to sell their shares on the market more quickly and at greater discounts than would be the case with the stock of a seasoned issuer.
The following factors also may add to the volatility in the price of our common stock: actual or anticipated variations in our quarterly or annual operating results; announcements regarding our clinical trials and the development and manufacture of our Hemopurifier; acceptance of our proprietary technology as a viable method of augmenting the immune response of clearing viruses and toxins from human blood; government regulations, announcements of significant acquisitions, strategic partnerships or joint ventures; our capital commitments and additions or departures of our key personnel. Many of these factors are beyond our control and may decrease the market price of our common shares regardless of our operating performance. We cannot make any predictions or projections as to what the prevailing market price for our common shares will be at any time, including as to whether our common shares will sustain their current market prices, or as to what effect the sale of shares or the availability of common shares for sale at any time will have on the prevailing market price.
Our issuance of additional shares of common stock or convertible securities could be dilutive.
We are entitled under our articles of incorporation to issue up to 60,000,000 shares of common stock. As of March 31, 2025, we have reserved for issuance 319,518 shares of common stock for outstanding restricted stock units, stock options and warrants, excluding an aggregate of 71,432 issuances of restricted stock units to our independent directors under our 2020 Equity Incentive Plan made subsequent to March 31, 2025. As of March 31, 2025, we had issued and outstanding 2,585,239 shares of common stock. As a result, as of March 31, 2025 we had 57,023,811 shares of common stock available for issuance to new investors or for use to satisfy indebtedness or pay service providers.
| 44 |
On March 16, 2025, Aethlon Medical, Inc. (the “Company”) entered into an inducement offer to exercise existing Class A and Class B Warrants (the “Agreement”) with a certain accredited and institutional holder (the “Holder”) of the Company’s outstanding Class A and Class B Warrants issued on May 17, 2024 (the “Existing Warrants”). Pursuant to the Agreement, the Holder, upon exercise, will receive a new unregistered Common Stock Purchase Warrant (“New Warrant”) pursuant to Section 4(a)(2) of the Securities Act of 1933, as amended (“Securities Act”), to purchase up to a number of shares equal to 200% of the number of Warrant Shares issued pursuant to the exercise of Existing Warrants pursuant to this Agreement (the “New Warrant Shares”), which New Warrant shall have an exercise price per share equal to $0.3736, subject to adjustment as provided in the New Warrant, will be exercisable at any time on or after six (6) months from the date of issuance and have a term of exercise of five and one-half (5.5) years from the date of issuance and a reduction of the exercise price of the Existing Warrants to $0.3736 per share, representing the closing price on March 14, 2025, but only with respect to a cash exercise under the Existing Warrants (as reduced from the current respective exercise price per share as set forth in the Existing Warrants).
The closing took place on March 17, 2025. Gross proceeds to the Company from the exercise of the Existing Warrants was $2,316,320, prior to deducting closing costs and placement agent fees as further described below. The Company intends to use the net proceeds from the offering for working capital and general corporate purposes.
As a result of the Holder exercising the Existing Warrants, the Company issued an aggregate of 6,200,000 shares of its common stock. The shares underlying the Existing Warrants have all been registered on Form S-1 registration statement (Registration Number 333-278188).
The Company agreed to file a resale registration statement registering the shares underlying the Replacement Warrants (“Resale Registration Statement”) within ninety (90) days of the date of the Agreement and to use commercially reasonable best efforts to cause the Resale Registration Statement to be effective on or prior to the 150th calendar day after the date of the Agreement.
Subject to the terms of the Agreement, the Company will be required to pay certain liquidated damages if the shares underlying the New Warrants are not filed within the ninety (90) period, as more fully described in the Agreement.
The Company further agreed that until sixty (60) days after the closing date of the warrant exercise, it will not (other than in connection with limited enumerated exceptions) issue, enter into any agreement to issue or announce the issuance or proposed issuance of any shares of common stock or common stock equivalents or file any registration statement or any amendment or supplement (other than the registration statement registering the shares underlying the Replacement Warrants).
In connection with the transactions contemplated in the Agreement, the Company agreed to pay its placement agent, Maxim Group, LLC (the “Agent”) the following compensation, (i) a cash fee equal to 6.0% of the gross proceeds received by the Company in the transactions contemplated by the Agreement, and (ii) legal fees and out-of-pocket expenses of $15,000.
On May 17, 2024, we closed a public offering pursuant to which we sold an aggregate of: (i) 2,450,000 shares of our common stock and accompanying Class A warrants to purchase up to 2,450,000 shares of common stock and Class B warrants to purchase up to 2,450,000 shares of common stock, at a combined public offering price of $0.58 per share and accompanying warrants; and (ii) in lieu of common stock, pre-funded warrants to purchase 5,650,000 shares of common stock and accompanying Class A warrants to purchase up to 5,650,000 shares of common stock and Class B warrants to purchase up to 5,650,000 shares of common stock, at a combined public offering price of $0.579 per pre-funded warrant and accompanying warrants, which is equal to the public offering price per share of common stock, and accompanying warrants less the $0.001 per share exercise price of each such pre-funded warrant.
Our Board of Directors may generally issue shares of common stock, restricted stock units or stock options or warrants to purchase those shares, without further approval by our stockholders, based upon such factors as our Board of Directors may deem relevant at that time. It is likely that we will be required to issue a large amount of additional securities to raise capital to further our development. It is also likely that we will be required to issue a large amount of additional securities to directors, officers, employees and consultants as compensatory grants in connection with their services, both in the form of stand-alone grants or under our stock plans.
| 45 |
Our officers and directors are entitled to indemnification from us for liabilities under our articles of incorporation, which could be costly to us and may discourage the exercise of stockholder rights.
Our articles of incorporation provide that we possess and may exercise all powers of indemnification of our officers, directors, employees, agents and other persons and our bylaws also require us to indemnify our officers and directors as permitted under the provisions of the Nevada Revised Statutes, or NRS. We may also have contractual indemnification obligations under our agreements with our directors, officers and employees. The foregoing indemnification obligations could result in our company incurring substantial expenditures to cover the cost of settlement or damage awards against directors and officers. These provisions and resultant costs may also discourage our company from bringing a lawsuit against directors, officers and employees for breaches of their fiduciary duties, and may similarly discourage the filing of derivative litigation by our stockholders against our directors, officers and employees even though such actions, if successful, might otherwise benefit our company and stockholders.
Our bylaws and Nevada law may discourage, delay or prevent a change of control of our company or changes in our management, would have the result of depressing the trading price of our common stock.
Certain anti-takeover provisions of Nevada law could have the effect of delaying or preventing a third-party from acquiring us, even if the acquisition arguably could benefit our stockholders.
Nevada’s “combinations with interested stockholders” statutes (NRS 78.411 through 78.444, inclusive) prohibit specified types of business “combinations” between certain Nevada corporations and any person deemed to be an “interested stockholder” for two years after such person first becomes an “interested stockholder” unless the corporation’s board of directors approves the combination (or the transaction by which such person becomes an “interested stockholder”) in advance, or unless the combination is approved by the board of directors and sixty percent of the corporation’s voting power not beneficially owned by the interested stockholder, its affiliates and associates. Further, in the absence of prior approval certain restrictions may apply even after such two year period. However, these statutes do not apply to any combination of a corporation and an interested stockholder after the expiration of four years after the person first became an interested stockholder. For purposes of these statutes, an “interested stockholder” is any person who is (1) the beneficial owner, directly or indirectly, of ten percent or more of the voting power of the outstanding voting shares of the corporation, or (2) an affiliate or associate of the corporation and at any time within the two previous years was the beneficial owner, directly or indirectly, of ten percent or more of the voting power of the then outstanding shares of the corporation. The definition of the term “combination” is sufficiently broad to cover most significant transactions between a corporation and an “interested stockholder.” A Nevada corporation may elect in its articles of incorporation not to be governed by these particular laws, but if such election is not made in the corporation’s original articles of incorporation, the amendment (1) must be approved by the affirmative vote of the holders of stock representing a majority of the outstanding voting power of the corporation not beneficially owned by interested stockholders or their affiliates and associates, and (2) is not effective until 18 months after the vote approving the amendment and does not apply to any combination with a person who first became an interested stockholder on or before the effective date of the amendment. We did not make such an election in our original articles of incorporation and have not amended our articles of incorporation to so elect.
Nevada’s “acquisition of controlling interest” statutes (NRS 78.378 through 78.3793, inclusive) contain provisions governing the acquisition of a controlling interest in certain Nevada corporations. These “control share” laws provide generally that any person that acquires a “controlling interest” in certain Nevada corporations may be denied voting rights, unless a majority of the disinterested stockholders of the corporation elects to restore such voting rights. These laws would apply to us if we were to have 200 or more stockholders of record (at least 100 of whom have addresses in Nevada appearing on our stock ledger) and do business in the State of Nevada directly or through an affiliated corporation, unless our articles of incorporation or bylaws in effect on the tenth day after the acquisition of a controlling interest provide otherwise. These laws provide that a person acquires a “controlling interest” whenever a person acquires shares of a subject corporation that, but for the application of these provisions of the NRS, would enable that person to exercise (1) one fifth or more, but less than one third, (2) one third or more, but less than a majority or (3) a majority or more, of all of the voting power of the corporation in the election of directors. Once an acquirer crosses one of these thresholds, shares which it acquired in the transaction taking it over the threshold and within the 90 days immediately preceding the date when the acquiring person acquired or offered to acquire a controlling interest become “control shares” to which the voting restrictions described above apply. These laws may have a chilling effect on certain transactions if our articles of incorporation or bylaws are not amended to provide that these provisions do not apply to us or to an acquisition of a controlling interest, or if our disinterested stockholders do not confer voting rights in the control shares.
| 46 |
Various provisions of our bylaws may delay, defer or prevent a tender offer or takeover attempt of us that a stockholder might consider in his or her best interest. Our bylaws may be adopted, amended or repealed by the affirmative vote of the holders of at least a majority of our outstanding shares of capital stock entitled to vote for the election of directors, and except as provided by Nevada law, our Board of Directors shall have the power to adopt, amend or repeal the bylaws by a vote of not less than a majority of our directors. The interests of these stockholders and directors may not be consistent with your interests, and they may make changes to the bylaws that are not in line with your concerns.
Nevada law also provides that directors may resist a change or potential change in control if the directors determine that the change is opposed to, or not in the best interests of, the corporation. The existence of the foregoing provisions and other potential anti-takeover measures could limit the price that investors might be willing to pay in the future for shares of our common stock. They could also deter potential acquirers of our company, thereby reducing the likelihood that you could receive a premium for your common stock in an acquisition.
We incur substantial costs as a result of being a public company and our management expects to devote substantial time to public company compliance programs.
As a public company, we incur significant legal, insurance, accounting and other expenses, including costs associated with public company reporting. We intend to invest resources to comply with evolving laws, regulations and standards, and this investment will result in increased general and administrative expenses and may divert management’s time and attention from product development and commercialization activities. If our efforts to comply with new laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to practice, regulatory authorities may initiate legal proceedings against us, and our business may be harmed. These laws and regulations could make it more difficult and costly for us to obtain director and officer liability insurance for our directors and officers, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These factors could also make it more difficult for us to attract and retain qualified executive officers and qualified members of our Board of Directors, particularly to serve on our audit and compensation committees. In addition, if we are unable to continue to meet the legal, regulatory and other requirements related to being a public company, we may not be able to maintain the quotation of our common stock on the Nasdaq Capital Market or on any other senior market to which we may apply for listing, which would likely have a material adverse effect on the trading price of our common stock.
If securities or industry analysts do not publish research or reports about our business, or if they change their recommendations regarding our stock adversely, our stock price and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that industry or securities analysts publish about us or our business. Our research coverage by industry and financial analysts is currently limited. Even if our analyst coverage increases, if one or more of the analysts who cover us downgrade our stock, our stock price would likely decline. If one or more of these analysts cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause our stock price or trading volume to decline.
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 1C. CYBERSECURITY
The Company’s Chief Financial Officer, with assistance from our third-party cybersecurity vendors, is responsible for identifying, assessing, and managing our cybersecurity threats and risks. Together, they identify and assess risks from cybersecurity threats by monitoring and evaluating our threat environment using various methods including, for example, automated tools, evaluating our and our industry’s risk profile, conducting real-time monitoring of certain systems, and implementing escalation protocols with our third-party cybersecurity vendors.
| 47 |
Depending on the environment, we implement and maintain various technical, physical, and organizational measures, processes, standards and policies designed to manage and mitigate material risks from cybersecurity threats to our Information Systems and Data, including, for example: an incident response plan; incident detection and response tools; encryption of certain sensitive data and certain data on mobile systems; certain access controls enforcing the principle of need-to-know encryption of certain sensitive data and certain data on mobile systems; physical security measures; asset management, tracking and disposal; monitoring of certain systems; and employee training.
Our assessment and management of material risks from cybersecurity threats are integrated into our overall risk management processes. For example, cybersecurity risk is addressed through our quality management system and processes and overseen by our audit committee of the board of directors. We use third-party service providers to assist us from time to time to identify, assess, and manage material risks from cybersecurity threats, including, for example, professional services firms, including legal counsel and certain cybersecurity service providers.
| 48 |
ITEM 2. PROPERTIES
Office, Lab and Manufacturing Space Leases
In December 2020, we entered into an agreement to lease approximately 2,823 square feet of office space and 1,807 square feet of laboratory space located at 11555 Sorrento Valley Road, Suite 203, San Diego, California 92121 and 11575 Sorrento Valley Road, Suite 200, San Diego, California 92121, respectively. The agreement carries a term of 63 months and we took possession of the office space effective October 1, 2021. We took possession of the laboratory space effective January 1, 2022. In October 2021, we entered into another lease for approximately 2,655 square feet of space to house our manufacturing operations located at 11588 Sorrento Valley Road, San Diego, California 92121. The term is for 55 months and we took possession of the manufacturing space in August 2022. The current monthly base rent under the office and laboratory component of the lease is $14,590. The current monthly base rent under the manufacturing component of the lease is $12,824.
The office, lab and manufacturing leases are coterminous with a remaining term of 24 months. The weighted average discount rate is 4.25%.
As of March 31, 2025, we have a right-of-use lease asset of $601,846.
The following table presents a maturity analysis of expected undiscounted cash flows for operating leases on an annual basis for the next two fiscal years. All of our leases conterminously expire during the fiscal year ending March 31, 2027.
| Fiscal Year Ended March 31, | |||
| 2026 | $ | 333,462 | |
| 2027 | 343,353 | ||
| Total minimum lease payments | 676,817 | ||
| Less amount representing imputed interest | (27,064 | ) | |
| Present value of minimum lease payments | $ | 649,751 | |
ITEM 3. LEGAL PROCEEDINGS
We may be involved from time to time in various claims, lawsuits, and/or disputes with third parties or breach of contract actions incidental to the normal course of our business operations. We are currently not involved in any litigation or any pending legal proceedings.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
| 49 |
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock is traded on the Nasdaq Capital Market under the trading symbol “AEMD.” On July 7, 2015, The Nasdaq Stock Market LLC approved our application for listing our common stock on the Nasdaq Capital Market under the symbol “AEMD,” and we commenced trading on the Nasdaq Capital Market on July 13, 2015. Previously, our common stock was quoted on the OTCQB Marketplace under the trading symbol “AEMD.”
Holders of Record
There were approximately 49 record holders of our common stock at June 24, 2025. The number of registered stockholders includes any beneficial owners of common shares held in street name.
Dividend Policy
We have not paid any dividends on our common stock to date and do not anticipate that we will pay dividends in the foreseeable future. Any payment of cash dividends on our common stock in the future will be dependent upon the amount of funds legally available, our earnings, if any, our financial condition, our anticipated capital requirements and other factors that the Board of Directors may think are relevant. However, we currently intend for the foreseeable future to follow a policy of retaining all of our earnings, if any, to finance the development and expansion of our business and, therefore, do not expect to pay any dividends on our common stock in the foreseeable future.
Recent Sales of Unregistered Securities
The Company did not have any sales of unregistered securities during the period covered by this Annual Report, other than those disclosed in our Form 8-K filed on March 17, 2025, which were exempt from registration under Section 4(a)(2) of the Securities Act.
Securities Authorized for Issuance Under Equity Compensation Plans
Information about our equity compensation plans is incorporated herein by reference to Item 12 of Part III of this Annual Report.
ITEM 6. [RESERVED]
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Overview
The following discussion and analysis should be read in conjunction with the consolidated Financial Statements and Notes thereto appearing elsewhere in this Annual Report.
We are a medical therapeutic company focused on developing the Hemopurifier® (HP), a clinical-stage immunotherapeutic device intended for applications in cancer, life-threatening viral infections, and organ transplantation and other areas of significant unmet needs. In human studies (164 sessions with 38 patients), the Hemopurifier was used safely and demonstrated the potential to remove enveloped viruses. In pre-clinical studies, the Hemopurifier has exhibited the capacity to remove harmful extracellular vesicles (EVs) and enveloped viruses from biological fluids, utilizing its proprietary lectin-based mechanism. These extracellular vesicles have been implicated in disease processes such as immune suppression and metastasis in cancer as well as in the progression of severe life-threatening infectious diseases. The U.S. Food and Drug Administration (“FDA”) has designated the Hemopurifier as a “Breakthrough Device” for two independent indications:
| · | the treatment of individuals with advanced or metastatic cancer who are unresponsive to or intolerant of standard of care therapy, and with cancer types in which extracellular vesicles have been shown to participate in the development or severity of the disease; and | |
| · |
the treatment of life-threatening viruses for which no approved therapies currently exist.
|
We are also evaluating the Hemopurifier’s potential in additional clinical contexts based on its mechanism of action and preclinical findings.
| 50 |
Oncology
We believe that the Hemopurifier may be a substantial advancement in the treatment of patients with advanced and metastatic cancer through its design to bind to and remove harmful remove harmful extracellular vesicles particles that promote the growth and spread of tumors. In October 2022, we formed a wholly-owned subsidiary in Australia to initially conduct oncology-related clinical research, then seek regulatory approval and commercialize our Hemopurifier in Australia.
We completed an in vitro binding study of extracellular vesicles from cancer patient samples, to provide pre-clinical evidence to support our trial design and translational endpoints. Our study indicated positive results from this study, providing evidence that our Hemopurifier removes extracellular vesicles, or EVs, from plasma. This translational study provides pre-clinical evidence to support our phase 1 safety, feasibility and dose-finding clinical trials of our Hemopurifier in patients with solid tumors who have stable or progressive disease during anti-PD-1 monotherapy treatment, such as Keytruda® or Opdivo®.
We have launched in an Australia safety, feasibility and dose-finding clinical trials of the Hemopurifier in cancer patients with solid tumors who have stable or progressive disease during anti-PD-1 monotherapy treatment, such as Keytruda® (pembrolizumab) or Opdivo® (nivolumab). The primary endpoint of the approximately nine to 18-patients, is safety. Exploratory analyses will be conducted to explore the number of HP treatments required to produce sustained reductions of EVs as well as improve anti-tumor T cell activity. We plan to open a similarly designed trial in India.
The following three hospitals in Australia have received ethics committee approval, have gone through training on our device and are open for patient enrollment: Royal Adelaide Hospital in Adelaide, Australia and Pindara Private Hospital in the Gold Coast section of Australia and GenesisCare North Shore Hospital in Sydney, Australia. As of 16JUN2025 we have treated three participants in the first of the three treatment cohorts. Once these patients have completed the pre-specified 7-day safety follow-up period, the data will be presented to an independent Data Safety Monitoring Board (DSMB). The DSMB will provide a recommendation to Aethlon senior leadership on advancing to the next cohort where participants will receive 2 HP treatments during the one week treatment period.
The Company continues to pursue approval of a similar clinical trial in India. HREC approval has previously been obtained at Medanta Medicity Hospital. Following this a meeting with Subject Expert Committee (SEC) of the India Regulatory Agency CDSCO was held 5JUN2025. We are awaiting the formal approval letter of the CDSCO. The clinical trial at Medanta can commence following a Site Initiation Visit (SIV) by the company’s India CRO, Qualtran.
Life-Threatening Viral Infections
We also believe that the Hemopurifier can be part of the broad-spectrum treatment of life-threatening highly glycosylated, or carbohydrate coated, viruses that are not addressed with an already approved treatment. In small-scale or early feasibility human studies, the Hemopurifier has been used in the past to treat individuals infected with human immunodeficiency virus, or HIV, hepatitis-C and Ebola.
Additionally, in vitro, the Hemopurifier has been demonstrated to capture Ebola, Marburg virus, Zika, Lassa, MERS-CoV, Cytomegalovirus, Epstein-Barr, Herpes simplex, Chikungunya, Dengue, West Nile, H1N1 swine flu, H5N1 bird flu, and the reconstructed 1918 Spanish flu virus. In several cases, these studies were conducted in collaboration with leading government or non-government research institutes.
The Hemopurifier has previously been studied under FDA and international regulatory frameworks for the treatment of severe SARS-CoV-2 infection. While we terminated our U.S. and India-based COVID-19 studies due to low ICU patient volume and shifting priorities, these programs demonstrated real-world use of the Hemopurifier in critically ill patients. We maintain an open IDE for viral indications to preserve optionality for future outbreaks or emergent pathogens.
We have sufficient inventory of Hemopurifiers to support our ongoing oncology trial in Australia as well as any near-term expansion of that study or potential trial activity in India. While we have received FDA approval to begin manufacturing at our San Diego facility under our IDE supplement, we are still awaiting FDA approval of a separate supplement to qualify an additional supplier of a key Hemopurifier component. We continue to work with the FDA on this process.
| 51 |
Pre-Clinical Exploration of Additional Clinical Uses for the Hemopurifier
The Aethlon R&D laboratory continues to explore potential new indications for the Hemopurifier. We have published in the peer-reviewed journal Transplant Immunology the ability of the device to remove extracellular vesicles and their microRNA cargo from acellular perfusates of discarded kidneys that had undergone normothermic machine perfusion.
On May 12, 2025, the results of our pre-clinical ex vivo study entitled “Ex Vivo Removal of CD41 positive platelet microparticles from Plasma by a Medical Device containing a Galanthus nivalis agglutinin (GNA) affinity resin” were published in the pre-print vehicle bioRxiv. This manuscript has been submitted to a peer-reviewed publication for review.
Platelet -derived extracellular vesicles (PD-EVs) are the most numerous EV population in the body and are released by platelets in response to a variety of stimuli. The cargo contained within these EVs have been noted to take part in damage to blood vessels, activation of immune cells and spread of tumor cells. Excessive levels of PD-EVs have been implicated in a myriad of diseases including cancer, lupus, systemic sclerosis, multiple sclerosis, Alzheimer’s disease, sepsis, acute and Long COVID.
We hypothesized that the Aethlon Hemopurifier which contains a propriety GNA affinity resin would remove platelet derived EVs from plasma. In this experiment two hundred milliliters on donated healthy human plasma were circulated over the Aethlon Hemoupurifier (HP) to simulate a clinical HP session. The study results showed a 98.5% removal of platelet -derived EVs at a timepoint equivalent to a 4-hour HP treatment. The results of this study support the current Australian Clinical Trial in Oncology as well as open the investigation of the Hemopurifier in many indications.
Extracellular vesicles have been implicated in the pathogenesis of Long COVID.As we had previously demonstrated removal of extracellular vesicles by the Hemopurifier in a patient with severe acute COVID-19 infection, we hypothesized that patients with Long COVID would have extracellular vesicles with the mannose sugar on their surface that would bind to the affinity resin in our device. We partnered with investigators at the Univ of California San Francisco Medical Center Long COVID clinic to obtain samples from participants with Long COVID as well as controls that had had COVID -10 infection but had recovered. The data to be presented will review the binding of larger and smaller extracellular vesicles to the GNA lectin and the lectin affinity resin, respectively. We believe the data from this pre-clinical study calls for additional study of the Hemopurifier and look forward to receiving feedback from the Long COVID scientific community at the Keystone Symposium.
Successful outcomes of human trials will also be required by the regulatory agencies of certain foreign countries where we plan to market and sell the Hemopurifier. Some of our patents may expire before FDA approval or approval in a foreign country, if any, is obtained. However, we believe that certain patent applications and/or other patents issued to us more recently will help protect the proprietary nature of our Hemopurifier treatment technology.
In addition to the foregoing, we are monitoring closely the impact of inflation, recent bank failures and the war between Russia and Ukraine and the military conflicts in Israel and the surrounding areas, as well as related political and economic responses and counter-responses by various global factors on our business. Given the level of uncertainty regarding the duration and impact of these events on capital markets and the U.S. economy, we are unable to assess the impact on our timelines and future access to capital. The full extent to which inflation, recent bank failures and the ongoing military conflicts will impact our business, results of operations, financial condition, clinical trials and preclinical research will depend on future developments, as well as the economic impact on national and international markets that are highly uncertain.
Our executive offices are located at 11555 Sorrento Valley Road, Suite 203, San Diego, California 92121. Our telephone number is (619) 941-0360. Our website address is www.aethlonmedical.com. The information contained on, or that can be accessed through, our website is not part of, and is not incorporated into, this Annual Report.
Our common stock is listed on the Nasdaq Capital Market under the symbol “AEMD.”
| 52 |
Fiscal Years Ended March 31, 2025 and 2024
Results of Operations
Government Contract Revenues
For the fiscal years ended March 31, 2025 and 2024, we did not have any active revenue-generating government contracts and, consequently, did not record any government contract revenue for that period.
Operating Costs and Expenses
Consolidated operating expenses were $9,341,364 for the fiscal year ended March 31, 2025, compared to $12,636,568 for the fiscal year ended March 31, 2024, a decrease of $3,295,203. The $3,295,203 decrease in the fiscal year ended March 31, 2025 was due to a decrease in payroll and related expenses of $1,332,359, a decrease of $1,302,834 in professional fees and a decrease of $660,010 in general and administrative expenses.
Payroll and related expenses decreased by $1,332,359 for the fiscal year ended March 31, 2025, compared to the prior year. The decrease was driven by a $876,511 reduction in salaries and related expenses and a $804,136 decrease in stock-based compensation. The reduction in salary expense reflects the termination of three executives—one in the prior year, one in July 2024, and one in October 2024—as well as a workforce reduction of non-executive employees implemented in August 2024. The decrease in stock-based compensation was primarily due to the absence of accelerated vesting charges recognized in the prior year in connection with the termination of our former Chief Executive Officer, as well as lower stock-based compensation expense associated with the departure of executives and non-executive staff. The overall decrease was partially offset by an increase of $348,287 in severance expenses mostly related to the termination of two former executives.
Professional fees decreased by $1,302,834 for the fiscal year ended March 31, 2025, compared to the prior year. The decrease was primarily driven by a $553,377 reduction in legal fees related to the transition to a new legal firm, a $462,154 decrease primarily attributable to the termination of services with a contract manufacturing organization and the completion of a project involving outside laboratory services. In addition, consulting fees related to scientific projects and regulatory projects decreased by $239,640 and $125,478 respectively. These decreases were partially offset by $84,900 increase in accounting fees associated with obtaining consents from prior audit firm for various SEC filings.
General and administrative expenses decreased by $660,010 for the fiscal year ended March 31, 2025, compared to the prior year. The decrease was primarily driven by a $534,069 reduction in costs related to lower purchases of raw materials for the production of Hemopurifiers, reduced cleanroom certification expenses, and fewer outside services for maintenance of the manufacturing facility. Laboratory supplies and testing costs also declined by $337,109 following the completion of oncology and transplant-related projects. Insurance expenses decreased by $141,453, including reductions in medical and workers’ compensation premiums due to lower headcount, as well as overall decrease in business insurance costs. Additional decreases included $44,122 in travel and entertainment expenses, $24,356 decrease in office supplies and $19,498 in depreciation expense related to the disposal of certain equipment. These decreases were partially offset by a $466,661 increase in clinical trial expenses related to our ongoing oncology study in Australia.
As a result of the above factors, our operating loss decreased to $9,341,364 for the fiscal year ended March 31, 2025, from $12,636,568 for the fiscal year ended March 31, 2024.
Other Income (Expense)
Other expense for the year ended March 31, 2025, included a non-cash charge of $4,612,862 related to a warrant inducement offer. In March 2025, the Company offered certain warrant holders the opportunity to exercise existing warrants at a temporarily reduced exercise price in exchange for the issuance of new warrants. The inducement expense recognized represents the combined fair value of the new warrants issued and the incremental fair value resulting from the modification of the exercise price of the existing warrants. This transaction did not impact cash flows from operating activities.
| 53 |
During the fiscal year ended March 31, 2025, we recognized $324,450 in other income related to the Employee Retention Tax Credit (“ERTC”) under the CARES Act and subsequent legislation. We recorded the ERTC as other income in the periods in which the payments were received. In addition, we recognized $36,339 in interest income related to the ERTC during fiscal 2025. As of March 31, 2025, the remaining expected credit was recorded as another receivable within other current assets on our consolidated balance sheet. No amounts were recorded in the prior fiscal year.
Liquidity and Capital Resources
As of March 31, 2025, we had a cash balance of $5,501,261 and working capital of $4,050,514. This compares to a cash balance of $5,441,978 and working capital of $4,395,889 at March 31, 2024.
While the Company has been carrying out certain expense reductions since November 2023; our planned additional expense reductions may not materialize and/or our patient recruitment may occur more rapidly than expected along with the concomitant increases in expenses, therefore there is substantial doubt that our cash on hand will carry the company for 12 months beyond the filing date of the financial statements included in this Annual Report.
During the fiscal year ended March 31, 2025 we raised capital through a warrant inducement offer and a public offering. In the fiscal year ended March 31, 2024 we raised money through our then existing At The Market Offering Agreement, or the 2022 ATM Agreement, with H.C. Wainwright & Co., LLC, or Wainwright. In October 2024, the S-3 registration underlying our At The Market Offering Agreement expired and the ATM was cancelled.
Financings During the Fiscal Year Ended March 31, 2025:
During the fiscal year ended March 31, 2025, we raised aggregate net proceeds of $7,746,311, net of $405,002 in commission and legal expenses to Maxim and $753,090 in direct legal and accounting fees. This total included $3,539,907 from a public offering in May 2024 and $2,054,940 from subsequent exercise of Class A and Class B warrants and $2,151,464 in net proceeds from a warrant inducement offering in March 2025. In connection with that transaction, we paid the placement agent fees totaling $153,979, consisting of a 6% commission on gross proceeds of $138,979 and $15,000 for legal and out-of-pocket expenses to Maxim and $10,877 in direct legal fees.
On March 16, 2025, Aethlon Medical, Inc. (the “Company”) entered into a inducement offer to exercise existing Class A and Class B Warrants (the “Agreement”) with a certain accredited and institutional holder (the “Holder”) of the Company’s outstanding Class A and Class B Warrants issued on May 17, 2024 (the “Existing Warrants”). Pursuant to the Agreement, the Holder, upon exercise, will receive a new unregistered Common Stock Purchase Warrant (“New Warrant”) pursuant to Section 4(a)(2) of the Securities Act of 1933, as amended (“Securities Act”), to purchase up to a number of shares equal to 200% of the number of Warrant Shares issued pursuant to the exercise of Existing Warrants pursuant to this Agreement (the “New Warrant Shares”), which New Warrant shall have an exercise price per share equal to $0.3736, subject to adjustment as provided in the New Warrant, will be exercisable at any time on or after six (6) months from the date of issuance and have a term of exercise of five and one-half (5.5) years from the date of issuance and a reduction of the exercise price of the Existing Warrants to $0.3736 per share, representing the closing price on March 14, 2025, but only with respect to a cash exercise under the Existing Warrants (as reduced from the current respective exercise price per share as set forth in the Existing Warrants).
The closing took place on March 17, 2025. Gross proceeds to the Company from the exercise of the Existing Warrants was $2,316,320, prior to deducting closing costs and placement agent fees as further described below. The Company intends to use the net proceeds from the offering for working capital and general corporate purposes.
As a result of the Holder exercising the Existing Warrants, the Company issued an aggregate of 775,000 shares of its common stock. The shares underlying the Existing Warrants have all been registered on Form S-1 registration statement (Registration Number 333-278188).
The Company agreed to file a resale registration statement registering the shares underlying the Replacement Warrants (“Resale Registration Statement”) within ninety (90) days of the date of the Agreement and to use commercially reasonable best efforts to cause the Resale Registration Statement to be effective on or prior to the 150th calendar day after the date of the Agreement.
| 54 |
Subject to the terms of the Agreement, the Company will be required to pay certain liquidated damages if the shares underlying the New Warrants are not filed within the ninety (90) period, as more fully described in the Agreement.
The Company further agreed that until sixty (60) days after the closing date of the warrant exercise, it will not (other than in connection with limited enumerated exceptions) issue, enter into any agreement to issue or announce the issuance or proposed issuance of any shares of common stock or common stock equivalents or file any registration statement or any amendment or supplement (other than the registration statement registering the shares underlying the Replacement Warrants).
In connection with the transactions contemplated in the Agreement, the Company agreed to pay its placement agent, Maxim Group, LLC (the “Agent”) the following compensation, (i) a cash fee equal to 6.0% of the gross proceeds received by the Company in the transactions contemplated by the Agreement, and (ii) legal fees and out-of-pocket expenses of $15,000.
On May 17, 2024, we closed a public offering pursuant to which we sold an aggregate of: (i) 306,250 shares of our common stock and accompanying Class A warrants to purchase up to 306,250 shares of common stock and Class B warrants to purchase up to 306,250 shares of common stock, at a combined public offering price of $4.64 per share and accompanying warrants; and (ii) in lieu of common stock, pre-funded warrants to purchase 706,250 shares of common stock and accompanying Class A warrants to purchase up to 706,250 shares of common stock and Class B warrants to purchase up to 706,250 shares of common stock, at a combined public offering price of $4.63 per pre-funded warrant and accompanying warrants, which is equal to the public offering price per share of common stock, and accompanying warrants less the $0.001 per share exercise price of each such pre-funded warrant. The gross proceeds from the offering, before deducting the placement agent’s fees and other offering expenses, were approximately $4.7 million.
Financings During the Fiscal Year Ended March 31, 2024:
During the fiscal year ended March 31, 2024, we raised aggregate net proceeds of $1,322,383, net of $34,118 in commissions to Wainwright and $8,202 in other offering expense, through the sale of 37,011 shares of our common stock at an average price of $35.76 per share under the 2022 ATM Agreement.
2022 At The Market Offering Agreement with H.C. Wainwright & Co., LLC
On March 24, 2022, we entered into the 2022 ATM Agreement with Wainwright, which established an at-the-market equity program pursuant to which we may offer and sell shares of our common stock from time to time as set forth in the 2022 ATM Agreement. This agreement was terminated in October 2024.
The offering was registered under the Securities Act pursuant to our shelf registration statement on Form S-3 (Registration Statement No. 333-259909), as previously filed with the SEC and declared effective on October 21, 2021. We filed a prospectus supplement, dated March 24, 2022, with the SEC that provides for the sale of shares of our common stock, or the 2022 ATM Shares, having an aggregate offering price of up to $15,000,000, which was subsequently and most recently updated pursuant to our prospectus supplement, dated September 29, 2022, filed with the SEC that provides for the sale of 2022 ATM Shares having an aggregate offering price of up to $6,625,000. As of March 31, 2024, $5,302,617 of 2022 ATM Shares remained available for sale under the 2022 ATM Agreement.
Under the 2022 ATM Agreement, Wainwright may sell the 2022 ATM Shares by any method permitted by law and deemed to be an “at the market offering” as defined in Rule 415 promulgated under the Securities Act, including sales made directly on the Nasdaq Capital Market, or on any other existing trading market for the 2022 ATM Shares. In addition, under the 2022 ATM Agreement, Wainwright may sell the 2022 ATM Shares in privately negotiated transactions with our consent and in block transactions. Under certain circumstances, we may instruct Wainwright not to sell the 2022 ATM Shares if the sales cannot be effected at or above the price designated by us from time to time.
We are not obligated to make any further sales of the 2022 ATM Shares under the 2022 ATM Agreement. The offering of the 2022 ATM Shares pursuant to the 2022 ATM Agreement will terminate upon the termination of the 2022 ATM Agreement by Wainwright or us, as permitted therein.
The 2022 ATM Agreement contains customary representations, warranties and agreements by us, and customary indemnification and contribution rights and obligations of the parties. We agreed to pay Wainwright a placement fee of up to 3.0% of the aggregate gross proceeds from each sale of the 2022 ATM Shares. We also agreed to reimburse Wainwright for certain specified expenses in connection with entering into the 2022 ATM Agreement.
| 55 |
Material Cash Requirements
We expect our clinical trial expenses for the planned oncology trials in Australia and India to increase for the foreseeable future. While these increases are primarily related to trial activities, additional Hemopurifiers may also be manufactured to support the studies.
In addition, we have entered into leases for our headquarters, laboratory and manufacturing facilities. We expect our rent payments to continue to increase for the foreseeable future.
Future capital requirements will depend upon many factors, including progress with pre-clinical testing and clinical trials, the number and breadth of our clinical programs, the time and costs involved in preparing, filing, prosecuting, maintaining and enforcing patent claims and other proprietary rights, the time and costs involved in obtaining regulatory approvals, competing technological and market developments, as well as our ability to establish collaborative arrangements, effective commercialization, marketing activities and other arrangements. We expect to continue to incur increasing negative cash flows and net losses for the foreseeable future. We will continue to need to raise additional capital either through equity and/or debt financing for the foreseeable future.
As a result of global events, political changes, bank failures, actual or perceived changes in interest rates and economic inflation, the global credit and financial markets have experienced extreme volatility, including diminished liquidity and credit availability, declines in consumer confidence, declines in economic growth, increases in inflation and uncertainty about economic stability. There can be no assurance that further deterioration in credit and financial markets and confidence in economic conditions will not occur. If equity and credit markets deteriorate, it may make any necessary debt or equity financing more difficult to obtain, more costly and/or more dilutive. Any of these actions could materially harm our business, results of operations and future prospects.
While we currently has been carrying out certain expense reductions since November 2023; our planned additional expense reductions may not materialize and/or our patient recruitment may occur more rapidly than expected along with the concomitant increases in expenses; therefore there is substantial doubt that our cash on hand will carry the company for 12 months beyond the filing date of the financial statements included in this Annual Report.
We do plan to access the equity markets for additional capital, however, there can be no assurance that we will be able to access such additional capital.
Our ability to raise additional funds may be adversely impacted by potential worsening global economic conditions and disruptions to and volatility in the credit and financial markets in the United States, including due to bank failures, actual or perceived changes in interest rates and economic inflation, and worldwide resulting from macroeconomic factors. Because of the numerous risks and uncertainties associated with product development, we cannot predict the timing or amount of increased expenses and we may never be profitable or generate positive cash flow from operating activities.
Cash Flows
Cash flows from operating, investing and financing activities, as reflected in the accompanying Consolidated Statements of Cash Flows, are summarized as follows (in thousands):
| For the year ended | ||||||
| March 31, 2025 |
March 31, 2024 |
|||||
| Cash provided by (used in): | ||||||
| Operating activities | $ | (7,646 | ) | $ | (10,130 | ) |
| Investing activities | – | (251 | ) | |||
| Financing activities | 7,727 | 1,288 | ||||
| Effect of exchange rate on cash | (12 | ) | 2 | |||
| Net increase (decrease) in cash | $ | 69 | $ | (9,091 | ) | |
| 56 |
Net Cash Used in Operating Activities
We used cash in our operating activities due to our losses from operations. Net cash used in operating activities was approximately $7,646,000 in fiscal 2025, compared to net cash used in operating activities of approximately $10,130,000 in fiscal 2024, a decrease of approximately $2,484,000. The decrease in cash flows was primarily driven by a decrease of $2,869,000 in net loss after non-cash charges partially offset by a negative change in our working capital items of $385,000 mainly from the approximately $210,000 decrease in accounts payable and other current liabilities and approximately $299,000 decrease in due to related parties. These decreases were partially offset by a decrease of approximately $124,000 in prepaids.
Net Cash Used in Investing Activities
During the fiscal year ended March 31, 2025 we did not purchase equipment. For the year ended March 31, 2024, we purchased approximately $251,000 of equipment, respectively.
Net Cash from Financing Activities
Net cash generated from financing activities increased from approximately $1,288,000 in the fiscal year ended March 31, 2024 to approximately $7,277,000 in the fiscal year ended March 31, 2025.
In the fiscal year ended March 31, 2025, we raised approximately $3,540,000 from the issuance of common stock. We also raised approximately $2,055,000 from the exercise of warrants under standard terms and approximately $2,316,000 from the exercise of warrants under induced terms. These proceeds were partially offset by $154,000 in commissions and $11,000 in legal fees related to the induced warrant exercises. Additionally, we used approximately $19,000 used to satisfy tax withholding obligations associated with the issuance of restricted stock units (RSUs).
During the fiscal year ended March 31, 2024, we raised $1,322,383 from the issuance of common stock, which was partially offset by the use of approximately $35,000 to pay for the tax withholding on the issuance of restricted stock units, or RSUs.
Critical Accounting Policies and Significant Judgments and Estimates
The preparation of consolidated financial statements in conformity with accounting principles generally accepted in the United States of America, or GAAP, requires us to make a number of estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements. These estimates and assumptions affect the reported amounts of expenses during the reporting period. On an ongoing basis, we evaluate estimates and assumptions based upon historical experience and various other factors and circumstances. We believe our estimates and assumptions are reasonable in the circumstances; however, actual results may differ from these estimates under different future conditions.
We believe that the estimates and assumptions that are most important to the portrayal of our financial condition and results of operations, in that they require the most difficult, subjective or complex judgments, form the basis for the accounting policies deemed to be most critical to us.
There were no accounting estimates in the year ended March 31, 2025 with a high degree of uncertainty or amounts that are with a high likelihood to change from period to period that would materially impact the presentation of our financial statements for the year ended March 31, 2025.
| 57 |
Warrant Inducement Transactions
From time to time, the Company may enter into warrant inducement arrangements, in which modifications to the terms of outstanding equity-classified warrants—such as reductions in exercise price or the issuance of additional warrants—are offered to incentivize early exercise. These transactions require significant judgment in determining whether the arrangement constitutes a routine equity modification or a substantive inducement that should be accounted for as an expense. In making this determination, the Company evaluates the structure and purpose of the transaction, including whether incremental value was transferred to the holder to accelerate capital inflows. In cases where the substance of the arrangement reflects an inducement, the Company records the incremental value as an expense in the period the transaction occurs. Determining the fair value of such inducements and the appropriate timing of recognition involves complex estimates and careful consideration of the facts and circumstances of each arrangement.
Share-based Compensation
We account for share-based compensation awards using the fair-value method and record such expense based on the grant date fair value in the consolidated financial statements over the requisite service period. This requires management to make estimates and assumptions regarding the fair value of the awards, including the expected term, volatility, risk-free interest rate, and forfeiture rates. These assumptions are inherently subjective and involve significant judgment. The fair value of stock options is typically determined using the Black-Scholes option pricing model. Compensation expense is recognized over the vesting period of the awards in a manner that reflects the service period or any applicable performance conditions.
RSU Grants to Non-Employee Directors
The Company maintains the Amended and Restated Non-Employee Director Compensation Policy, or the Director Compensation Policy, which provides for cash and equity compensation for persons serving as non-employee directors of the Company. Under this policy, each new director receives either stock options or a grant of RSUs upon appointment/election, as well as either an annual grant of stock options or of RSUs at the beginning of each fiscal year. The (i) stock options are subject to vesting and (ii) RSUs are subject to vesting and represent the right to be issued on a future date shares of our common stock upon vesting.
On April 16, 2024, our Board of Directors approved, pursuant to the terms of the Director Compensation Policy, the grant of the annual RSUs under the Director Compensation Policy to each of the four non-employee directors of the Company then serving on the Board of Directors. The Director Compensation Policy provides for a grant of stock options or $50,000 worth of RSUs at the beginning of each fiscal year for current non-employee directors then serving on the Board of Directors, and for a grant of stock options or $75,000 worth of RSUs for a newly elected non-employee director, with each RSU priced at the average for the closing prices for the five days preceding and including the date of grant, or $12.16 per share for the RSUs granted in April 2024. As a result, in April 2024 the four eligible directors were each granted an RSU in the amount of 4,112 shares under the Company’s 2020 Equity Incentive Plan, or the 2020 Plan. The RSUs are subject to vesting in four equal installments, with 25% of the restricted stock units vesting on each of June 30, 2024, September 30, 2024, December 31, 2024, and March 31, 2025, subject in each case to the director’s Continuous Service (as defined in the 2020 Plan), through such dates. Vesting will terminate upon the director’s termination of Continuous Service prior to any vesting date.
There were no vested RSUs outstanding as of March 31, 2025.
Recent Events
Reverse Split – Following the approval of a reverse stock split at a Special Meeting of Stockholders on May 13, 2025, our Board of Directors approved a 1-for-8 reverse stock split or our outstanding shares of Common Stock, effective as of the close of business on June 6, 2023. Accordingly, each 8 shares of outstanding common stock held by stockholders were combined into one share of common stock. Our authorized common stock remained at 60,000,000 shares following the stock split. We issued 77 additional shares shares as a result of rounding up fractional shares related to the reverse stock split.
| 58 |
On June 2, 2025, a second patient was treated with the Hemopurifier at GenesisCare North Shore Hospital in Sydney, Australia. The patient was treated with the Aethlon Hemopurifier for 4 hours in a single day and tolerated the procedure without complications. The patient will have follow-up safety visits, EV and T cell measurements as well as imaging for clinical response.
RSU Grants
In April 2025, our Board of Directors approved, pursuant to the terms of the Director Compensation Policy, the grant of the annual RSUs under the Director Compensation Policy to each of the four non-employee directors of the Company then serving on the Board of Directors. The Director Compensation Policy provides for a grant of stock options or $50,000 worth of RSUs at the beginning of each fiscal year for current non-employee directors then serving on the Board of Directors, and for a grant of stock options or $75,000 worth of RSUs for a newly elected non-employee director, with each RSU priced at the average for the closing prices for the five days preceding and including the date of grant, or $2.80 per share for the April 2024 RSU grants. As a result, in April 2025 the four eligible directors were each granted an RSU in the amount of 17,858 shares under the 2020 Plan. The RSUs are subject to vesting in four equal installments, with 25% of the restricted stock units vesting on each of June 30, 2025, September 30, 2025, December 31, 2025, and March 31, 2026, subject in each case to the director’s Continuous Service (as defined in the 2020 Plan), through such dates. Vesting will terminate upon the director’s termination of Continuous Service prior to any vesting date.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Not applicable to a “smaller reporting company” as defined under Item 10(f)(1) of Regulation S-K of the Securities Act.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
| Pages | |
| Report of Independent Registered Public Accounting Firm (Haskell & White, LLP, San Diego, CA PCAOB ID No. 200) | F-2 |
| Report of Independent Registered Public Accounting Firm (Baker Tilly US, LLP, San Diego, CA PCAOB ID No. 23) | F-4 |
| Consolidated Balance Sheets | F-5 |
| Consolidated Statements of Operations | F-6 |
| Consolidated Statements of Equity | F-7 |
| Consolidated Statements of Cash Flows | F-8 |
| Notes to Consolidated Financial Statements | F-9 |
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
| 59 |
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
We maintain “disclosure controls and procedures” (as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act) that are designed to ensure that information required to be disclosed, in our Exchange Act reports is recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our Interim Chief Executive Officer and Chief Financial Officer (who is our principal executive officer and principal financial officer), to allow timely decisions regarding required disclosures.
In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management is required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures. We have carried out an evaluation as of the end of the period covered by this Annual Report under the supervision and with the participation of our management, including our Chief Executive Officer, who also serves as our Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures.
Internal Control over Financial Reporting
| (a) | Management’s Report on Internal Control Over Financial Reporting |
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Under the supervision and with the participation of our management, including our Chief Executive Officer, who also serves as our Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of March 31, 2025. The evaluation was conducted in accordance with the guidelines established by the Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Based on this evaluation, and the successful implementation of remediation measures addressing the previously identified material weaknesses, management concluded that our internal control over financial reporting was effective as of March 31, 2025.
Remediation of Previously Reported Material Weaknesses
Segregation of Duties and User Access Controls
As previously disclosed, management identified a material weakness in internal control over financial reporting related to segregation of duties and user access controls within our financial systems. Specifically, the Company had not adequately maintained user access controls to restrict both user and privileged access to financial applications, allowing the same individual to initiate, record, and approve accounting entries. Additionally, check stock was stored in the office of an authorized signatory, representing a weakness in physical access control.
This control deficiency was due in part to our limited accounting and finance staff, which hindered the design and implementation of effective segregation of duties. The lack of adequate user access controls and the absence of sufficient compensating controls could have resulted in a material misstatement of the financial statements.
| 60 |
To remediate this material weakness, the Company implemented the following measures:
| · | In November 2023, check stock was relocated to a secure area accessible only to individuals without check signing authority, to mitigate unauthorized access risks. Subsequently, the Company discontinued maintaining any physical check stock onsite. While the Company continues to issue physical checks as needed, these are processed exclusively through the bank’s controlled check issuance services, eliminating the risk associated with onsite check stock and enhancing control over disbursements. | |
| · | In May 2024, the Company completed an upgrade to its accounting software, enabling the establishment of distinct user roles and system access controls. While certain individuals may both initiate and record transactions due to the size of the finance team, all transactions are reviewed and approved by personnel independent of those entering the transactions. Management has tested the design and operating effectiveness of these new controls and concluded that the material weakness was remediated as of March 31, 2025. |
Accounting for Accrued Commission Liabilities
The Company also previously identified a material weakness in internal control over financial reporting related to the accounting for accrued commission liabilities. From fiscal 2017 through fiscal 2020, the Company incorrectly recorded commission accruals totaling approximately $404,000. This error arose due to the incorrect application of U.S. GAAP and the absence of adequate review controls, and it was not identified and corrected in a timely manner.
The Company reversed the erroneous accrued commission liability during the year ended March 31, 2024, with the correction reflected as a reclassification to equity. To prevent recurrence, the Company implemented quarterly controls during fiscal 2025 for the review and validation of accruals, including those related to commissions. These controls include formal procedures for the assessment of accrual balances, with overall oversight provided by the Audit Committee in connection with its review of the Company’s financial reporting.
Management has evaluated the design and operating effectiveness of these controls and concluded that the material weakness was remediated as of March 31, 2025.
Limitations on Internal Control Over Financial Reporting
Management recognizes that a system of internal control over financial reporting, no matter how well designed and operated, can provide only reasonable assurance and may not prevent or detect all material misstatements. Internal control over financial reporting has inherent limitations, including the possibility of human error, the circumvention or overriding of controls, or changes in conditions that may render controls inadequate. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions or because the degree of compliance with the policies or procedures may deteriorate.
| (b) | Changes in Internal Control Over Financial Reporting |
There was no change in our internal control over financial reporting during the last fiscal quarter ended March 31, 2025 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
During the three
months ended March 31, 2025, none of our directors or officers
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
| 61 |
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The names, ages and positions of our directors and executive officers as of June 26, 2025 are listed below:
| NAMES | TITLE OR POSITION(1) | AGE | ||
| James B. Frakes | Chief Executive Officer, Chief Financial Officer and Director | 68 | ||
| Edward G. Broenniman | Chairman and Director | 88 | ||
| Angela Rossetti | Director | 71 | ||
| Chetan S. Shah, M.D. | Director | 55 | ||
| Nicolas Gikakis | Director | 58 | ||
| Steven P. LaRosa, M.D. | Chief Medical Officer | 58 |
| (1) | Our Board of Directors has determined that Mr. Broenniman, Mr. Gikakis, Ms. Rossetti and Dr. Shah meet the requirements to be determined as “independent directors” for all purposes, including Compensation Committee and Audit Committee purposes, under the Nasdaq Stock Market (“Nasdaq”) rules and for federal securities law purposes. Mr. Frakes is not independent, as he also functions as executive and officer of the Company. |
Certain additional information concerning the individuals named above is set forth below. This information is based on information furnished to us by each individual noted.
James B. Frakes Chief Executive Officer, Chief Financial Officer and Director
Mr. Frakes has served as Chief Executive Officer and Chief Financial Officer since October 3, 2024, having previously served as Interim Chief Executive Officer beginning in November 2023. He has also served as a director of the Company since November 2023, and has held the role of Chief Financial Officer of the Company since September 2010. Prior to being appointed as Chief Financial Officer, Mr. Frakes served as Senior Vice President, Finance of the Company from January 2008 to September 2010. He previously served as the Chief Financial Officer for Left Behind Games Inc., a start-up video game company. Prior to 2006, he served as Chief Financial Officer of NTN Buzztime, Inc., an interactive entertainment company. Mr. Frakes received an MBA from the University of Southern California and a B.A. with Honors from Stanford University.
Edward G. Broenniman, Chairman and Director
Mr. Broenniman has served as a director of the Company since March 1999. He has been the managing director of The Piedmont Group, LLC, a venture advisory firm, since 1978. Mr. Broenniman currently serves on the boards of two privately held firms. He previously served on the boards of the nonprofit entities, the Dingman Center for Entrepreneurship’s Board of Advisors at the University of Maryland (1989 to 2020), the National Capital Chapter of Corporate Directors (Founder, Chair from 2003 to 2005 and director from 2001 to 2018) and the Board of the Association for Corporate Growth, National Capital Chapter (Founder, Chair from 2000 to 2018). Mr. Broenniman received his MBA from Stanford Graduate School of Business and his B.A. from Yale University.
| 62 |
Nicolas Gikakis, Director
Mr. Gikakis has served as a director of the Company since July 2023. From 2021 to May 2023, Mr. Gikakis served as the Head of Commercial for WearOptimo Pty Ltd, a private Australian medical device and digital health company. Previously, from 2017 to 2019, Mr. Gikakis served as Vice President of Strategy and Corporate Development at Oventus Medical Limited, a private medical device company, during which time he assisted with the commercial expansion of its sleep apnea device. From 2012 to 2021, Mr. Gikakis held various leadership and independent strategic advisor positions in the healthcare industry in sales, marketing, product development, and corporate development and transactions, including for companies working with blood filtration and purification. Mr. Gikakis earned a B.S. in bioengineering from the University of Pennsylvania and holds an MBA from George Mason University, with earlier work in bench and clinical research, and clinical experience at the University of Pennsylvania.
Angela Rossetti, Director
Ms. Rossetti has served as a director of the Company since April 2022. As an active consultant since March 2018, her client list has included Kala Pharmaceuticals, Inc. and Celgene Corporation, among others. From June 2015 through July 2017, Ms. Rossetti served as Vice President of Cell Machines, Inc., an early-stage biopharmaceutical company developing novel protein therapies, where she assisted with the commercialization of technology for hemophilia and other diseases. Ms. Rossetti has held a number of positions within pharmaceutical commercial development, marketing, communications and finance, including Vice President of a Global Commercial Medicine Team at Pfizer Inc. from 2007 to 2012, where she led a global smoking cessation campaign. Ms. Rossetti previously served on the board of directors of Palatin Technologies, Inc., a public biopharmaceutical company, from June 2013 to December 2020. Ms. Rossetti currently holds positions as an adjunct Assistant Professor of Medical and Pharmaceutical Ethics at New York Medical College and an Adjunct Associate at Albert Einstein College of Medicine. Ms. Rossetti graduated from a joint program of the Albert Einstein College of Medicine and Benjamin N. Cardozo School of Law with an M.S. in Bioethics, has an M.B.A. from Columbia University Graduate School of Business and a B.A. in Biology and English from the University of Pennsylvania.
Chetan S. Shah, M.D., Director
Dr. Shah has served as a director of the Company since June 2013. Dr. Shah is a board certified Otolaryngologist. He is a partner and board member of the Surgery Center at Hamilton, as well as Physician Management Systems and Princeton Eye & Ear, which he founded in 2009. Dr. Shah serves on the board of one other private company. He holds teaching positions and serves on multiple hospital committees in the area and is on the Audiology and Speech Language Pathology Committee for the State of New Jersey. Dr. Shah also was a member of the Board of Medical Examiners for the State of New Jersey. Dr. Shah received his Bachelor’s degree and Medical Degree from Rutgers University and Robert Wood Johnson Medical School, respectively.
Steven P. LaRosa, M.D., Chief Medical Officer
Dr. LaRosa has served as our Chief Medical Officer since January 2021 and served as our Chief Scientific Officer from May 2021 until February 2023. Dr. LaRosa has over 20 years of experience as a practicing physician and infectious disease specialist. Prior to joining the Company, Dr. LaRosa served as the Vice President of Clinical Development of Entasis Therapeutics, a spin-out of AstraZeneca focused on pathogen-targeted small molecules to treat serious multidrug-resistant Gram-negative infections, from March 2020 to December 2020. Prior to joining Entasis, Dr. LaRosa was an Attending Physician in the Division of Infectious Disease at Beverly Hospital, a member of Beth Israel Lahey Health. Prior to Beverly Hospital from September 2012 to March 2020, he was an Attending Physician in the Division of Infectious Diseases at Rhode Island Hospital. Prior to that, Dr. LaRosa was an Associate Staff Physician in the Department of Infectious Disease at the Cleveland Clinic Foundation. He also served as a Clinical Research Physician for Eli Lilly and Company. Throughout his career, Dr. LaRosa has had several academic appointments. Dr. LaRosa holds his M.D. from Boston University School of Medicine and his B.S. in Biology from Boston College. He completed an Internal Medicine Residency and Chief Residency at the Cleveland Clinic Foundation and Infectious Disease Fellowship at Massachusetts General Hospital. He is Board Certified by the ABIM in Internal Medicine and Infectious Disease.
| 63 |
Family Relationships
There are no family relationships between or among the directors or executive officers.
There are no arrangements or understandings between any two or more of our directors or executive officers or between any of our directors or executive officers and any other person pursuant to which any director or officer was or is to be selected as a director or officer, and there is no arrangement, plan or understanding as to whether non-management stockholders will exercise their voting rights to continue to elect the current Board of Directors. There are also no arrangements, agreements or understandings between non-management stockholders that may directly or indirectly participate in or influence the management of our affairs.
Legal Proceedings
To our knowledge, (i) no director or executive officer has been a director or executive officer of any business that has filed a bankruptcy petition or had a bankruptcy petition filed against it during the past ten years; (ii) no director or executive officer has been convicted of a criminal offense or is the subject of a pending criminal proceeding during the past ten years; (iii) no director or executive officer has been the subject of any order, judgment or decree of any court permanently or temporarily enjoining, barring, suspending or otherwise limiting his involvement in any type of business, securities or banking activities during the past ten years; and (iv) no director or officer has been found by a court to have violated a federal or state securities or commodities law during the past ten years.
Board of Directors
Our Board of Directors has the responsibility for establishing broad corporate policies and for overseeing our overall performance. Members of our Board of Directors are kept informed of our business activities through discussions with our Interim Chief Executive Officer and other executive officers, by reviewing analyses and reports sent to them and by participating in Board and committee meetings. Mr. Broenniman serves as Chairman of our Board and Mr. Frakes as our Interim Chief Executive Officer, and we have not designated a lead independent director. We believe that having the offices of Chairman of our Board and Interim Chief Executive Officer held by two different people is appropriate for a company of our size and stage of development in order to maximize efficiencies of our limited available personnel resources. Nevada law provides that each director holds office after the expiration of his or her term until a successor is elected and qualified, or until the director resigns or is removed, resulting in a term that extends to our next annual meeting of stockholders. Our Board of Directors presently has an Audit Committee, a Compensation Committee and a Nominating and Corporate Governance Committee, on which each of Mr. Broenniman and Ms. Rossetti serve as independent directors. In addition, Dr. Shah serves as an independent director on the Audit, Compensation and Nominating and Corporate Governance Committees, and Mr. Gikakis serves as an independent director on the Nominating and Corporate Governance Committees. Mr. Broenniman is Chair of the Audit Committee, Dr. Shah is Chair of the Compensation Committee and Ms. Rossetti is Chair of the Nominating and Corporate Governance Committee.
Our Board of Directors believes that sound governance practices and policies provide an important framework to assist them in fulfilling their duty to stockholders. Our Board of Directors has implemented separate committees for the areas of audit, compensation and nomination of directors, annual review of the independence of our Audit and Compensation Committee members, maintenance of a majority of independent directors and written expectations of management and directors, among other best practices.
Our Board of Directors has determined that four of our five current directors meet the independence requirements of the Nasdaq Capital Market, on which our common stock is listed. In the judgment of our Board of Directors, Mr. Frakes does not meet such independence standards, as he serves as an executive officer of the Company. In reaching its conclusions, our Board of Directors considered all relevant facts and circumstances with respect to any direct or indirect relationships between our Company and each of the directors, including those discussed under the caption “Certain Relationships and Related Transactions,” below. Our Board of Directors determined that any relationships that exist or existed in the past between our Company and each of the independent directors were immaterial on the basis of the information set forth in the above-referenced sections.
| 64 |
Audit Committee and Audit Committee Financial Expert
Our Board of Directors formed an Audit Committee in May 1999. Our Board of Directors has determined that Mr. Broenniman, due to his professional experience business acumen and independence, meets the definition of an “audit committee financial expert” as defined in Item 407(d)(5)(ii) under Regulation S-K, promulgated under the Exchange Act.
Each of the members of the Audit Committee has a basic understanding of finance and accounting and is able to read and understand fundamental financial statements. Our Board of Directors has determined that each of the members of the Audit Committee meets the independence requirements applicable to audit committee members of Nasdaq Capital Market companies. The Audit Committee has the authority to appoint, review and discharge our independent registered public accounting firm. The Audit Committee reviews the results and scope of the audit and other services provided by our independent registered public accounting firm, as well as our accounting principles and our system of internal controls, reports the results of their review to the full Board of Directors and to management and recommends to the full Board of Directors that our audited consolidated financial statements be included in our Annual Report on Form 10-K.
The Audit Committee has adopted a charter, which can be found on our website under “Investors – Governance – Governance Documents.” The reference to or inclusion of our website address in this Amendment No. 1 does not include or incorporate by reference the information on our website into this Amendment No. 1.
Compensation Committee
The Compensation Committee approves or makes recommendations to our Board of Directors on decisions concerning compensation of the executive management team and non-employee directors and administers our stock-based incentive compensation plans. The Chair establishes meeting agendas after consultation with other committee members. Our Interim Chief Executive Officer and other members of management regularly discuss our compensation issues with Compensation Committee members. Subject to Compensation Committee review, modification and approval, our Chief Executive Officer typically makes recommendations respecting bonuses and equity incentive awards for the other members of the executive management team. The Compensation Committee establishes all bonus and equity incentive awards for all executive members of the management team. Our Board of Directors has determined that all members of the Compensation Committee meet the independence requirements applicable to Nasdaq Capital Market companies.
With respect to calendar year 2024, our Compensation Committee considered compensation information provided by Anderson Pay Advisors LLC (“Anderson”), a compensation consultant, in determining executive compensation. Anderson provided competitive compensation data showing that our cash compensation generally was and made cash compensation recommendations designed to compensate our officers in line with the 50% range for similarly situated companies.
The Compensation Committee has adopted a charter, which can be found on our website at “Investors –Governance – Governance Documents.” The reference to or inclusion of our website address in this Amendment No. 1 does not include or incorporate by reference the information on our website into this Amendment No. 1.
Nominating and Corporate Governance Committee
The responsibilities of the Nominating and Corporate Governance Committee include:
| · | overseeing our corporate governance functions on behalf of our Board of Directors; | |
| · | making recommendations to our Board of Directors regarding corporate governance issues; | |
| · | identifying and evaluating candidates to serve as directors of our Company consistent with criteria approved by our Board of Directors; | |
| · | selecting director candidates or recommending such candidates to our Board of Directors for selection; and | |
| · | reviewing and evaluating the performance of our Board of Directors. |
| 65 |
The Nominating and Corporate Governance Committee has adopted a charter, which can be found on our website at “Investors – Governance – Governance Documents.” The reference to or inclusion of our website address in this Amendment No. 1 does not include or incorporate by reference the information on our website into this Amendment No. 1.
Stockholder Nominees for Director
There have been no material changes to the procedures by which stockholders may recommend nominees to the Board of Directors.
Code of Ethics
In February 2005, our Board of Directors approved a “Code of Business Conduct and Ethics” (as amended from time to time, the “Code”), which applies to our principal executive officer, our principal financial officer, our principal accounting officer and persons performing similar tasks. In February 2020, the Board of Directors adopted an amended Code, which is applicable to all of our directors, officers and other employees and which is available on our website at www.aethlonmedical.com. If we make any substantive amendments to, or grant any waivers from, the Code for any officer or director, we will disclose the nature of such amendment or waiver on our website or in a Current Report on Form 8-K. The inclusion of our website address in this Amendment No. 1 does not include or incorporate by reference the information on our website into this Amendment No. 1.
Incentive Compensation Recoupment Policy
We have adopted an incentive compensation recovery policy (the “Compensation Recovery Policy”) that is designed to comply with, and will be interpreted in a manner consistent with, Section 10D and Rule 10D-1 of the Exchange Act and the applicable rules of the Nasdaq Stock Market, including any interpretive guidance provided by Nasdaq. Under our Compensation Recovery Policy, in the event of an accounting restatement due to the Company’s material noncompliance with any financial reporting requirement under the securities laws, including any required accounting restatement to correct a material error in previously issued financial statements, or that would result in a material misstatement if the error were corrected in the current period or left uncorrected in the current period, the Company must recover erroneously awarded incentive-based compensation previously paid to the Company’s executive officers in accordance with the terms of such Compensation Recovery Policy. Furthermore, under the Compensation Recovery Policy, the Company is prohibited from indemnifying any executive officer or former executive officer against the loss of erroneously awarded incentive-based compensation and from paying or reimbursing an executive officer for purchasing insurance to cover any such loss.
A copy of our Compensation Recovery Policy is attached as Exhibit 97.1 to this Amendment No. 1.
Delinquent Section 16(a) Reports
Section 16(a) of the Exchange Act requires the Company’s directors and executive officers and persons who beneficially own more than ten percent of a registered class of the Company’s equity securities to file with the Commission initial reports of ownership and reports of changes in ownership of Common Stock and other equity securities of the Company. Officers, directors and greater than ten percent beneficial stockholders are required by Commission regulations to furnish us with copies of all Section 16(a) forms they file. To the best of the Company’s knowledge based solely on a review of Forms 3, 4, and 5 (and any amendments thereof) received by us during or with respect to the year ended March 31, 2025 and written representations that no other reports were required, there were no late Section 16 filings during the year ended March 31, 2025.
ITEM 11. EXECUTIVE COMPENSATION
We are a “smaller reporting company” under Item 10 of Regulation S-K promulgated under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and the following compensation disclosure is intended to comply with the requirements applicable to smaller reporting companies. Although the rules allow us to provide less detail about our executive compensation program, the Compensation Committee of our Board of Directors (the “Compensation Committee”) is committed to providing the information necessary to help stockholders understand its executive compensation-related decisions. Accordingly, this section includes supplemental narratives that describe the 2024 fiscal year executive compensation program for our named executive officers.
| 66 |
Our named executive officers for the fiscal year ended March 31, 2025, consist of our Chief Executive Officer, who also serves as our Principal Financial Officer; our Chief Medical Officer; and our former Senior Vice President, Chief Operating Officer, who served during the fiscal year but was no longer employed as of year-end. These individuals represented our principal executive officers and the two most highly compensated executive officers other than our principal executive officers.
| · | James B. Frakes, our Chief Executive Officer and Chief Financial Officer; | |
| · | Steven P. LaRosa, M.D., our Chief Medical Officer; and | |
| · | Guy F. Cipriani, our former Senior Vice President, Chief Operating Officer. |
SUMMARY COMPENSATION TABLE FOR 2025 AND 2024 FISCAL YEARS
The following table summarizes all compensation earned by our named executive officers for the fiscal years ended March 31, 2025 and 2024.
| Named And Principal Position | Fiscal Year Ended March 31, |
Salary ($) |
All Other Compensation ($) |
Total ($) |
||||||||||||
| James B. Frakes | 2025 | 500,000 | – | 500,000 | ||||||||||||
| Chief Executive Officer and Chief Financial Officer | 2024 | 416,449 | – | 416,449 | ||||||||||||
| Steven P. LaRosa, M.D. | 2025 | 430,000 | 50,000 | 480,000 | ||||||||||||
| Chief Medical Officer | 2024 | 430,000 | – | 430,000 | ||||||||||||
| Guy F. Cipriani | 2025 | 199,500 | 196,188 | (1) | 395,688 | |||||||||||
| Senior Vice President, Chief Operating Officer | 2024 | 378,064 | – | 378,064 | ||||||||||||
| (1) | Includes a severance payment of $178,750 and a vacation payout of $17,438, each paid in connection with the executive’s termination of employment. |
Narrative Disclosure to Executive Summary
Generally, the three principal components of our executive compensation program for our named executive officers are base salary, executive cash bonus and long-term incentive equity compensation. We do not have any formal policies for allocating compensation among salary, performance bonus awards and equity grants, short-term and long-term compensation or among cash and non-cash compensation. Instead, the Compensation Committee considered compensation information provided by Anderson Pay Advisors LLC, or Anderson, our compensation consultant, in determining the compensation to recommend to the Board of Directors for its approval, that it believes appropriate to achieve the goals of our executive compensation program and our corporate objectives. We generally target providing total executive and director compensation at the 50% range for comparable companies.
Base Salary
Base salary provides financial stability and security to our named executive officers through a fixed amount of cash for performing job responsibilities. Each of our named executive officers’ 2025 and 2024 calendar year base salaries are listed in the table below, which reflects the Compensation Committees’ review of the data provided by Anderson and the Compensation Committee’s goal of setting salaries to be at the 50% range for comparable companies.
| Name | 2025 Base Salary | 2024 Base Salary | ||||||
| James B. Frakes | $ | 500,000 | $ | 500,000 | (1) | |||
| Steven P. LaRosa, M.D. | $ | 430,000 | $ | 430,000 | ||||
| Guy F. Cipriani | – | $ | 390,000 | (2) | ||||
| (1) | Mr. Frakes’ annual base salary was increased from $360,000 to $500,000, effective as of November 7, 2023 in connection with his appointment as interim Chief Executive Officer | |
| (2) | Mr. Cipriani’s annual base salary was increased from $370,000 to $390,000, effective as of November 7, 2023 in connection with his appointment as Senior Vice President, Chief Operating Officer. Mr. Cipriani’s employment was terminated on October 3, 2024. |
| 67 |
Executive Cash Bonuses and Annual Cash Incentives
With respect to the fiscal year ended March 31, 2025, we approved a cash bonus of $50,000 to our Chief Medical Officer. We did not approve any annual cash incentives for our named executive officers.
Equity-Based Incentive Awards
Individual stock option grants are determined based on a number of factors, including current corporate and individual performance, outstanding equity holdings and their retention value and total ownership, historical value of our stock, internal equity amongst executives and market data provided by Anderson. In the fiscal year ended March 31, 2025, we did not approve any equity-based incentive awards for our named executive officers.
Granting of Certain Equity Awards Close in Time to the Release of Material Nonpublic Information
Employment and Separation Agreements
On December 12, 2018, we entered into an executive employment agreement with Mr. Frakes, which was amended in November 2023 and currently governs the terms of his employment. Effective November 7, 2023, in connection with his appointment as Interim Chief Executive Officer, the Board of Directors increased Mr. Frakes’ annual base salary to $500,000. On October 3, 2024, Mr. Frakes was appointed as our permanent Chief Executive Officer. He will also continue to serve as Chief Financial Officer.
The agreement also provides that Mr. Frakes is eligible to receive an annual cash performance bonus, based on the achievement of individual and company performance objectives to be established annually by the Board of Directors or its Compensation Committee. The decision to award a bonus, and the amount, is at the discretion of the Board of Directors or the Compensation Committee.
In the event Mr. Frakes’ employment is terminated without cause or he resigns for good reason (as defined in the agreement), he will be entitled to continue receiving his base salary and company-paid healthcare premiums for 12 months following such termination.
Effective July 1, 2024, Lee Arnold, Chief Science Officer of the Company, was terminated by the Company. As a result of Mr. Cipriani’s termination, that executive employment agreement entered into by and between Mr. Arnold and the Company on February 1, 2023, (the “Employment Agreement”), terminated July 1, 2024. In connection with his departure, Mr. Arnold is entitled to receive twelve months’ severance and related separation benefits, consistent with the terms of his Employment Agreement.
Effective October 3, 2024, Guy F. Cipriani, Chief Operating Officer of the Company, has departed the Company. Mr. Cipriani’s departure was not related to the Company’s financial or operating results or to any disagreements or concerns regarding the Company’s financial or reporting practices.
As a result of Mr. Cipriani’s departure, that executive employment agreement entered into by and between Mr. Cipriani and the Company on January 2, 2021, as amended (the “Employment Agreement”), terminated effective October 3, 2024. In connection with his departure, Mr. Cipriani is entitled to receive twelve months’ severance and related separation benefits, consistent with the terms of his Employment Agreement.
On January 4, 2021, we entered into an executive employment agreement with Dr. LaRosa, which governs the current terms of his employment with us. Dr. LaRosa’s annual base salary was increased by the Compensation Committee to $430,000, effective May 1, 2021, when Dr. LaRosa assumed the additional duties of interim Chief Scientific Officer, which he held until February 2023. In addition, we paid Dr. LaRosa a one-time signing bonus of $100,000. Further, Dr. LaRosa was eligible to receive a grossed-up reimbursement of relocation expenses pursuant to the terms of his employment agreement. In addition, the agreement provides that Dr. LaRosa is eligible for an annual cash performance bonus for each year with a target amount of 40% of Dr. LaRosa’s then-current annual base salary, based upon our and Dr. LaRosa’s achievement of objectives and milestones to be determined on an annual basis by the Board of Directors (or Compensation Committee thereof). Whether Dr. LaRosa receives an annual bonus for any given year, and the amount of any such annual bonus, will be determined in the discretion of our Board of Directors (or the Compensation Committee thereof). The agreement also provides that if Dr. LaRosa’s employment is terminated without cause, or if he resigns for good reason (each as defined in the agreement), then Dr. LaRosa will be entitled under his agreement to continue to receive his annual base salary and payment of premiums for continuation of healthcare benefits for a period of 12 months following such termination.
| 68 |
Outstanding Equity Awards at 2025 Fiscal Year-End
The following table sets forth certain information concerning equity awards granted to our named executive officers that remained outstanding as of March 31, 2025.
| OPTIONS AWARDS | ||||||||||||||||||||
| Name | Grant Date | Number of Securities Underlying Unexercised Options Exercisable (#) |
Number of Securities Underlying Unexercised Options Unexercisable (#) |
Option Exercise Price ($) |
Option Expiration Date |
|||||||||||||||
| James B. Frakes | 4/3/2020 | 1,75620 | (1) | – | 102.40 | 4/2/2030 | ||||||||||||||
| Chief Executive Officer and | 2/10/2022 | 1,253 | (2) | 287 | 112.80 | 2/9/2032 | ||||||||||||||
| Chief Financial Officer | ||||||||||||||||||||
| Steven P. LaRosa, M.D. | 1/4/2021 | 1,512 | (3) | – | 201.60 | 1/3/2031 | ||||||||||||||
| Chief Medical Officer | 2/10/2022 | 1,253 | (4) | 287 | 112.80 | 2/9/2032 | ||||||||||||||
| Guy F. Cipriani, MBA, | – | – | – | – | – | |||||||||||||||
| Senior Vice President, Chief Operating Officer | – | – | – | – | – | |||||||||||||||
| (1) | This option is subject to vesting at a rate of 25% on the one year anniversary of the grant date of April 3, 2020, then monthly over the following 36 months, subject to Mr. Frakes continued service with the Company. | |
| (2) | This option is subject to vesting at a rate of 25% on the one year anniversary of the grant date of February 10, 2022, then monthly over the following 36 months, subject to Mr. Frakes continued service with the Company. | |
| (3) | This option is subject to vesting at a rate of 25% on the one year anniversary of the grant date of January 4, 2021, then monthly over the following 36 months, subject to Dr. LaRosa’s continued service with the Company. | |
| (4) | This option is subject to vesting at a rate of 25% on the one year anniversary of the grant date of February 10, 2022, then monthly over the following 36 months, subject to Dr. LaRosa’s continued service with the Company. | |
| (5) | This option is subject to vesting at a rate of 25% on the one year anniversary of the grant date of January 4, 2021, then monthly over the following 36 months, subject to Mr. Cipriani’s continued service with the Company. | |
| (6) | This option is subject to vesting at a rate of 25% on the one year anniversary of the grant date of February 10, 2022, then monthly over the following 36 months, subject to Mr. Cipriani’s continued service with the Company. |
| 69 |
Director Compensation for 2025 Fiscal Year
The following director compensation disclosure reflects all compensation awarded to, earned by or paid to our then non-employee directors for the fiscal year ended March 31, 2025.
| Fees Earned or Paid in Cash ($) |
Stock Awards ($)(1) |
Total ($) |
||||||||||
| Edward G. Broenniman (2) | 97,500 | 50,000 | 147,500 | |||||||||
| Nicolas Gikakis (3) | 48,750 | 50,000 | 98,750 | |||||||||
| Angela Rossetti (4) | 63,000 | 50,000 | 113,000 | |||||||||
| Chetan S. Shah, M.D. (5) | 63,750 | 50,000 | 113,750 | |||||||||
| (1) | In accordance with SEC rules, this column reflects the aggregate grant date fair value of the awards computed in accordance with Financial Accounting Standard Board Accounting Standards Codification Topic 718 for stock-based compensation transactions. Assumptions used in the calculation of these amounts are included in our consolidated financial statements in this Annual Report. These amounts do not reflect the actual economic value that will be realized by our directors upon the vesting, exercise, or the sale of the shares of common stock underlying such awards. | |
| (2) | In the fiscal year ended March 31, 2025, Mr. Broenniman earned $30,000 in cash compensation for his services to us as non-executive Chairman and $67,500 related to his roles as a member of our Audit Committee, Compensation Committee and Nominating and Corporate Governance Committee and as the chair of our Audit Committee, for an aggregate amount of $97,500. Mr. Broenniman also received restricted stock units, or RSUs, valued at $50,000 for his ongoing service as a Board member pursuant to our Amended and Restated Non-Employee Director Compensation Policy, or Director Compensation Policy. | |
| (3) | Mr. Gikakis served on our Audit Committee until September 30,2024. In the fiscal year ended March 31, 2025, Mr. Gikakis earned $48,750 for his roles as a director and as a member of our Audit Committee and Nominating and Corporate Governance Committee. Mr. Gikakis also received RSUs valued at $50,000 for his ongoing service as a Board member pursuant to our Director Compensation Policy. As of March 31, 2024, Mr. Gikakis had 4,885 shares of common stock subject to outstanding RSUs. | |
| (4) | In the fiscal year ended March 31, 2025, Ms. Rossetti earned $63,000 for her roles as a director, a member of our Audit Committee, Compensation Committee and Nominating and Corporate Governance Committee and as the chair of our Nominating and Corporate Governance Committee. Ms. Rossetti also received RSUs valued at $50,000 for her ongoing service as a Board member pursuant to our Director Compensation Policy. As of March 31, 2025, Ms. Rossetti had no outstanding equity awards. | |
| (5) | Dr. Shah served as a member of our Audit Committee until September 15, 2023. In the fiscal year ended March 31, 2025, Dr. Shah earned $63,750 for his roles as a director, a member of our Audit Committee, Compensation Committee and Nominating and Corporate Governance Committee and as the chair of our Compensation Committee. Dr. Shah also received RSUs valued at $50,000 for his ongoing service as a Board member pursuant to our Director Compensation Policy. |
Non-Employee Director Compensation Policy
We maintain the Director Compensation Policy, in which only non-employee directors may participate, pursuant to which such non-employee directors are entitled to receive cash and equity compensation for their service on the Board of Directors and its committees. Under the Director Compensation Policy in effect during the fiscal year ended March 31, 2025, a newly appointed or elected eligible director will receive an initial grant of RSUs with a grant date fair value of $75,000 or, at the discretion of our Board of Directors, options to acquire shares of common stock with a grant date fair value of $75,000, based on the average of the closing prices of our common stock for the five trading day period ending on the date of grant and will vest at a rate determined by the Board of Directors in its discretion, typically in equal quarterly installments over one year.
In addition, under the Director Compensation Policy, at the beginning of each fiscal year, each continuing director eligible to participate will receive a grant of RSUs with a grant date fair value of $50,000 or, at the discretion of our Board of Directors, options to acquire shares of common stock with a grant date fair value of $50,000, based on the average of the closing prices of our common stock for the five trading day period ending on the date of grant and will vest at a rate determined by the Board of Directors in its discretion, typically in equal quarterly installments over one year.
| 70 |
Under the Director Compensation Policy in effect during the fiscal year ended March 31, 2025, in lieu of per meeting fees, eligible directors will receive an annual board retainer fee of $40,000, as well as the following annual retainer fees: Audit Committee chair - $15,000, Compensation Committee chair - $15,000, Nominating and Corporate Governance Committee chair - $8,000, Audit Committee member - $7,500 (not applicable to the chair), Compensation Committee member - $7,500 (not applicable to the chair) and Nominating Committee member - $5,000 (not applicable to the chair). Additionally, the Chairperson of the Board of Directors will receive an additional annual board retainer fee of $30,000.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The following table sets forth information as of March 31, 2025, with respect to the ownership of our common stock, by (i) each person known by us to be the beneficial owner of more than five percent (5%) of the outstanding shares of each class of our capital stock, (ii) each of our directors and director nominees, (iii) each of our executive officers, and (iv) all of our named executive officers and directors as a group. As of such date, we had 2,585,316 shares of our common stock issued and outstanding, after adjustment for the 1-for-8 reverse stock split effective as of the close of business on June 6, 2025 with an effective trading date of June 9, 2025. We believe that each individual or entity named has sole investment and voting power with respect to shares of common stock indicated as beneficially owned by them, subject to community property laws where applicable, except where otherwise noted.
Unless otherwise indicated, the address for each person listed in the table below is c/o Aethlon Medical, Inc., 11555 Sorrento Valley Road, Suite 203, San Diego, CA 92121.
| NAME OF BENEFICIAL OWNER | NUMBER OF SHARES BENEFICIALLY OWNED(1) |
PERCENT OF SHARES BENEFICIALLY OWNED(2) |
||||||
| Greater than 5% Stockholders | ||||||||
| Armistice Capital, LLC | 180,758 | (3) | 7% | |||||
| Ikarian Capital, LLC | 153,660 | (4) | 6% | |||||
| Directors and Named Executive Officers | ||||||||
| James B. Frakes, Chief Executive Officer, Chief Financial Officer and Director | 2,805 | (5) | * | |||||
| Edward G. Broenniman, Chairman and Director | 4,887 | (6) | * | |||||
| Chetan S. Shah, M.D., Director | 3,923 | (7) | * | |||||
| Angela Rossetti, Director | 6,208 | (8) | * | |||||
| Steven P. LaRosa, M.D., Chief Medical Officer | 2,552 | (9) | * | |||||
| Nicolas Gikakis, Director | 3,569 | (10) | – | |||||
| All Current Directors and Executive Officers as a Group (6 members) | 23,944 | 9% | ||||||
____________________
| * | Less than 1% |
| (1) | Calculated pursuant to Rule 13d-3(d)(1) of the Exchange Act. Under Rule 13d-3(d)(1), shares not outstanding that are subject to options, warrants, rights or conversion privileges exercisable by a person within 60 days are deemed outstanding for the purpose of calculating the number and percentage owned by such person but not deemed outstanding for the purpose of calculating the percentage owned by each other person listed. | |
| (2) | Based on 2,585,316 shares of common stock outstanding as of March 31, 2025, as adjusted for the 1-for-8 reverse stock split effective as of the close of business on June 6, 2025 with an effective trading date of June 9, 2025. | |
| (3) | Armistice Capital, LLC ("Armistice Capital") is the investment manager of Armistice Capital Master Fund Ltd. (the "Master Fund"), the direct holder of the Shares, and pursuant to an Investment Management Agreement, Armistice Capital exercises voting and investment power over the securities of the Issuer held by the Master Fund and thus may be deemed to beneficially own the securities of the Issuer held by the Master Fund. Mr. Boyd, as the managing member of Armistice Capital, may be deemed to beneficially own the securities of the Issuer held by the Master Fund. The Master Fund specifically disclaims beneficial ownership of the securities of the Issuer directly held by it by virtue of its inability to vote or dispose of such securities as a result of its Investment Management Agreement with Armistice Capital. The Master Fund, a Cayman Islands exempted company that is an investment advisory client of Armistice Capital, has the right to receive dividends from, or the proceeds from the sale of, the reported securities. The address of the principal business office of each of the Reporting Persons is c/o Armistice Capital, LLC, 510 Madison Avenue, 7th Floor, New York, NY 10022. |
| 71 |
| (4) | Represents shares of common stock held by Ikarian Healthcare Master Fund, L.P., a Cayman Islands exempted limited partnership (the “Fund”) and certain separate managed account. The Fund, and certain separately managed accounts managed by Ikarian Capital (collectively, the "Managed Accounts"), are the record owners of the securities covered by this statement. Ikarian Capital is an investment adviser registered under the Investment Advisers Act of 1940, as amended, and serves as investment manager to the Fund and as sub-adviser to the Managed Accounts, and may be deemed to have beneficial ownership of the securities covered by this statement through the investment discretion it has over the Fund and the Managed Accounts. Ikarian Capital is ultimately controlled, indirectly, by Mr. Shahrestani. Accordingly, Mr. Shahrestani may be deemed to indirectly beneficially own securities beneficially owned by Ikarian Capital. The Fund disclaims beneficial ownership of the shares held by the Managed Accounts. The Managed Accounts disclaim beneficial ownership of the shares held by the Fund. The address of the principal business office of each of the Reporting Persons is c/o Ikarian Capital, LLC, 100 Crescent Court, Suite 1620, Dallas, Texas 75201. | |
| (5) | Consists of (i) 30 shares of common stock and (ii) 2,805 shares subject to stock options that are currently exercisable or will be exercisable within 60 days of March 31, 2025. | |
| (6) | Consist of 4,887 shares of common stock | |
| (7) | Consist of 3,923 shares of common stock. | |
| (8) | Consists of 6,208 shares of common stock. | |
| (9) | Consists of 2,552 shares subject to stock options that are currently exercisable or will be exercisable within 60 days of March 31, 2025. | |
| (10) | Consists of 3,569 shares of common stock |
Equity Compensation Plans
The following table sets forth information, as of March 31, 2025, about our equity compensation plans in effect as of that date:
| Plan category | (a) Number of securities to be issued upon exercise of outstanding options, warrants and rights (1) |
(b) Weighted-average exercise price of outstanding options, warrants and rights |
(c) Number of securities remaining available for future issuance under equity compensation plans (excluding securities reflected in column (a)) |
|||||||||
| Equity compensation plans approved by security holders (2) | 6,546 | $ | 130.52 | 390,950 | ||||||||
| Equity compensation plans not approved by security holders (3) | – | – | – | |||||||||
| Totals | 6,546 | $ | 130.52 | 390,950 | ||||||||
| (1) | Net of equity instruments forfeited, exercised or expired. | |
| (2) | Excludes RSU grants to our officers and directors during the fiscal year ended March 31, 2025, since all of the shares underlying the RSUs had been issued during that fiscal year and there were no outstanding RSUs as of March 31, 2025. | |
| (3) | As of March 31, 2025, we did not have any equity compensation plans that were not approved by our stockholders. |
| 72 |
Item 402(v) Pay Versus Performance
We are providing the following information about the relationship between executive compensation actually paid and certain financial performance of our company as required by Section 953(a) of the Dodd-Frank Wall Street Reform and Consumer Protection Act and Item 402(v) of Regulation S-K. The disclosure included in this section is prescribed by SEC rules and does not necessarily align with how the Company or the Compensation Committee view the link between the Company’s performance and named executive officer (“NEO”) pay. This disclosure is intended to comply with the requirements of Item 402(v) of Regulation S-K applicable to “smaller reporting companies.” For additional information about our compensation philosophy and how we seek to align executive compensation with the Company’s performance, refer to “Executive and Director Compensation” section above.
Required Tabular Disclosure of Pay Versus Performance
The amounts set forth below under the headings “Compensation Actually Paid to PEO” and “Average Compensation Actually Paid to Non-PEO NEOs” have been calculated in a manner consistent with Item 402(v) of Regulation S-K. Use of the term “compensation actually paid” is required by the SEC’s rules and as a result of the calculation methodology required by the SEC, such amounts differ from compensation actually received by the individuals and the compensation decisions described in the “Executive and Director Compensation” section above. With the exception of Charles J. Fisher as PEO during the 2023 fiscal year, our Chief Executive Officer is our principal executive officer and is referred to as PEO in the headers in the following tables.
| PAY VERSUS PERFORMANCE | ||||||||||||||||||||||||||||||||||
| Fiscal Year |
Summary Compensation Table Total for PEO 1(1) ($) |
Compensation Actually Paid to PEO 1(2)(4) ($) |
Summary Compensation Table Total for PEO 2(1) ($) |
Compensation Actually Paid to PEO 2(3)(4) ($) |
Average Summary Compensation Table Total for Non-PEO NEOs(5) ($) |
Average Compensation Actually Paid to Non-PEO NEOs(6) ($) |
Value of Initial Fixed $100 Investment Based On Total Shareholder Return(7) ($) |
Net Income (Loss) (millions)(8) ($) |
||||||||||||||||||||||||||
| (a) | (b) | (c) | (b) | (c) | (d) | (e) | (f) | (h) | ||||||||||||||||||||||||||
| 2025 | $ | – | $ | – | $ | 500,000 | $ | 495,402 | $ | 430,844 | $ | 430,875 | $ | 2.45 | $ | (13.40 | ) | |||||||||||||||||
| 2024 | $ | 505,332 | $ | 395,249 | $ | 416,449 | $ | 400,496 | $ | 404,032 | $ | 385,270 | $ | 8.28 | $ | (12.21 | ) | |||||||||||||||||
| 2023 | $ | 460,000 | $ | (62,027 | ) | – | – | $ | 388,750 | $ | 220,700 | $ | 18.84 | $ | (12.03 | ) | ||||||||||||||||||
| (1) | The dollar amounts reported in these columns (b) are the amounts of total compensation reported for our respective PEOs for each corresponding year in the “Total” column of the Summary Compensation Table. Refer to “Executive Compensation — Summary Compensation Table.” | |
| (2) | Charles J. Fisher, Jr., M.D. served as the Company’s Chief Executive Officer for the entirety of the year fiscal year ended March 31, 2023 and through November 7, 2023, and served as the Company’s PEO for such period. | |
| (3) | James B. Frakes was appointed as the Company’s Chief Executive Officer, effective November 7, 2023, and served as the Company’s PEO from such date through March 31, 2024. | |
| (4) | The dollar amounts reported in columns (c) represent the amount of “compensation actually paid” to Dr. Fisher and Mr. Frakes in the periods during which they served as PEO, respectively, as computed in accordance with Item 402(v) of Regulation S-K. The dollar amounts do not reflect the actual amount of compensation earned by or paid to our PEOs during the applicable year. In accordance with the requirements of Item 402(v) of Regulation S-K, the following adjustments were made to the reported total compensation for our PEOs for each year to determine the compensation actually paid: |
| 73 |
| PEO 1 | PEO 2 | |||||||||||||||||||||||
| 2025 ($) |
2024 ($) |
2023 ($) |
2025 ($) |
2024 ($) |
2023 ($) |
|||||||||||||||||||
| Summary Compensation Table Total | – | 505,332 | 460,000 | 500,000 | 416,449 | 388,750 | ||||||||||||||||||
| Deduct: Grant Date Fair Value of Equity Awards as reported in Summary Compensation Table (a) | – | – | – | – | – | |||||||||||||||||||
| Add: Fiscal Year-End Fair Value of Equity Awards Granted in the Fiscal Year | – | – | – | – | – | |||||||||||||||||||
| Add: Change in Fair Value of Outstanding and Unvested Equity Awards Granted in Prior Fiscal Years | – | – | (378,862 | ) | (9,739 | ) | (128,266 | ) | ||||||||||||||||
| Add: Fair Value of Equity Awards Granted in the Fiscal Year that Vested in the Fiscal Year | – | – | – | (2,465 | ) | – | – | |||||||||||||||||
| Add: Change in Fair Value as of the Vesting Date of Equity Awards Granted in Prior Fiscal Years that Vested in the Fiscal Year | – | – | (143,164 | ) | (2,133 | ) | (39,784 | ) | ||||||||||||||||
| Deduct: Fair Value at the End of the Prior Year of Equity Awards that Failed to Meet Vesting Conditions in the Year | – | (110,083 | ) | – | – | – | ||||||||||||||||||
| Compensation Actually Paid (b) | – | 395,249 | (62,027 | ) | 495,402 | 400,496 | 220,700 | |||||||||||||||||
| (a) | The grant date fair value of equity awards represents the total of the amounts reported in the “Option Awards” column in the Summary Compensation Table for the applicable year. | |
| (b) | Amount of equity award adjustments may differ from amount reported in the table above due to rounding. The fair values of stock options vested during the fiscal year or outstanding as of fiscal year end were estimated using the Black-Scholes option pricing model with the following assumptions, which may be materially different from the assumptions used for estimating the grant-date fair value as reported in the “Option Awards” columns in the Summary Compensation Table: |
| Year Ended March 31, | ||||||||||||
| 2025 | 2024 | 2023 | ||||||||||
| Expected term (in years) | 4.45 years | 4,46 years | 5.20 years | |||||||||
| Expected volatility | 137.13% | 145.9% | 138.9% | |||||||||
| Risk-free interest rate | 4.07% | 4.02% | 2.20% | |||||||||
| Expected dividend rate | 0.00% | 0.00% | 0.00% | |||||||||
| (5) | The dollar amounts reported in column (d) represent the average of the amounts reported for the NEOs as a group (excluding our PEOs) in the “Total” column of the Summary Compensation Table in each applicable year. The NEOs (excluding our PEO) included for purposes of calculating the average amounts in each applicable year are Dr. LaRosa and Mr. Cipriani. | |
| (6) | The dollar amounts reported in column (e) represent the average amount of “compensation actually paid” to our non-PEO NEOs, as computed in accordance with Item 402(v) of Regulation S-K. The dollar amounts do not reflect the actual amount of compensation earned by or paid to our NEOs during the applicable year. In accordance with the requirements of Item 402(v) of Regulation S-K, the following adjustments were made to the reported total compensation for each year to determine the compensation actually paid: |
| 74 |
| Average of Non-PEO NEOs | ||||||||||||
| 2025 | 2024 | 2023 | ||||||||||
| Summary Compensation Table Total | 437,844 | 404,032 | 388,750 | |||||||||
| Deduct: Grant Date Fair Value of Equity Awards as reported in Summary Compensation Table (a) | – | – | – | |||||||||
| Add: Fiscal Year-End Fair Value of Equity Awards Granted in the Fiscal Year | – | – | – | |||||||||
| Add: Change in Fair Value of Outstanding and Unvested Equity Awards Granted in Prior Fiscal Years | (1,232 | ) | (13,524 | ) | (128,266 | ) | ||||||
| Add: Fair Value of Equity Awards Granted in the Fiscal Year that Vested in the Fiscal Year | – | – | – | |||||||||
| Add: Change in Fair Value as of the Vesting Date of Equity Awards Granted in Prior Fiscal Years that Vested in the Fiscal Year | (3,002 | ) | (5,238 | ) | (39,784 | ) | ||||||
| Deduct: Fair Value at the End of the Prior Year of Equity Awards that Failed to Meet Vesting Conditions in the Year | (2,734 | ) | – | – | ||||||||
| Compensation Actually Paid (b) | (430,875 | ) | (385,270 | ) | (62,027 | ) | ||||||
| (a) | The grant date fair value of equity awards represents the total of the amounts reported in the “Option Awards” column in the Summary Compensation Table for the applicable year. | |
| (b) | Amount of equity award adjustments may differ from amount reported in the table above due to rounding. The fair values of stock options vested during the fiscal year or outstanding as of fiscal year end were estimated using the Black-Scholes option pricing model with the following assumptions, which may be materially different from the assumptions used for estimating the grant-date fair value as reported in the “Option Awards” columns in the Summary Compensation Table: |
| Year Ended March 31, | ||||||||||||
| 2025 | 2024 | 2023 | ||||||||||
| Expected term (in years) | 4.45 years | 4.46 years | 5.20 years | |||||||||
| Expected volatility | 137.13% | 145.9% | 138.9% | |||||||||
| Risk-free interest rate | 4.07% | 4.02% | 2.20% | |||||||||
| Expected dividend rate | 0.00% | 0.00% | 0.00% | |||||||||
| (7) | TSR is determined based on the value of an initial fixed investment of $100 on March 31, 2022. Cumulative TSR is calculated by dividing the sum of the cumulative amount of dividends for the measurement period, assuming dividend reinvestment, and the difference between the Company’s share price at the end and the beginning of the measurement period by the Company’s share price at the beginning of the measurement period. | |
| (8) | Net loss attributable to Aethlon as reported in the Company’s consolidated financial statements for the applicable year. |
| 75 |
Required Narrative Disclosure to Pay Versus Performance Table
In accordance with Item 402(v) of Regulation S-K, we are providing the following descriptions of the relationships between information presented in the Pay Versus Performance table above.
Compensation Actually Paid and Net Income Loss
Because the Company is a pre-commercial stage company, we had no revenue during the periods presented, other than revenue associated with government contracts and grants. Consequently, we do not use net income (loss) as a performance measure in our executive compensation program. Moreover, as a pre-commercial stage company with limited revenue, we do not believe there is any meaningful relationship between our net loss and compensation actually paid to our NEOs during the periods presented.
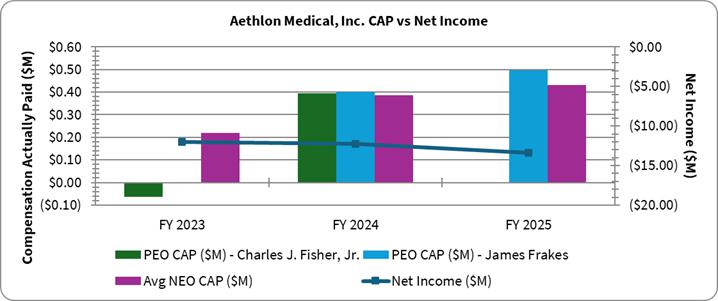
Compensation Actually Paid and Cumulative TSR
The chart below shows the relationship between the compensation actually paid to our PEOs and the average compensation actually paid to our non-PEO NEOs, on the one hand, to the Company’s cumulative TSR over the three years presented in the table, on the other.
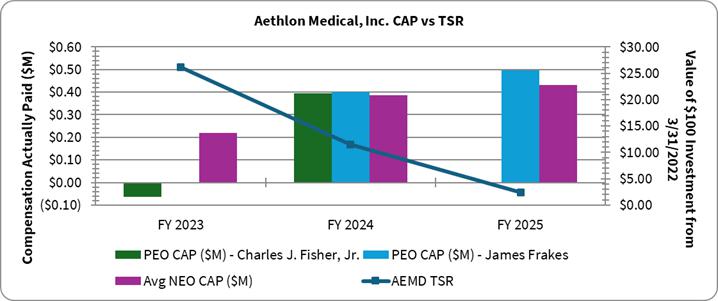
All information provided above under the “Item 402(v) Pay Versus Performance” heading will not be deemed to be incorporated by reference into any filing of the Company under the Securities Act of 1933, as amended, or the Securities Exchange Act of 1934, as amended, whether made before or after the date hereof and irrespective of any general incorporation language in any such filing, except to the extent the Company specifically incorporates such information by reference.
| 76 |
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS AND DIRECTOR INDEPENDENCE
The following describes all transactions since April 1, 2023, and all proposed transactions, in which we were or are to be a participant and the amount involved exceeds the lesser of $120,000 or one percent of the average of our total assets at year-end for the last two completed fiscal years, and in which any related person had or will have a direct or indirect material interest. In making such decisions our Audit Committee considers and approves or disapproves any related party transaction as defined under SEC Regulation Item 404, to the extent required by SEC regulations.
Separation Agreement with Former CEO
In connection with Charles J. Fisher, Jr. M.D.’s resignation as the Company’s Chief Executive Officer, effective November 2023, (the “Fisher Separation Date”), in accordance with the terms of his Executive Employment Agreement with the Company, dated as of October 30, 2020 (the “Fisher Employment Agreement”), and pursuant to Dr. Fisher’s Separation Agreement with the Company, effective as of November 27, 2023 (the “Fisher Separation Agreement”), the Company will provide Dr. Fisher with (1) cash severance equivalent to twelve months of Dr. Fisher’s base salary in effect as of the Separation Date, subject to standard payroll deductions and withholdings, payable over the Company’s regular payroll schedule over the twelve months following the Separation Date; (2) the accelerated vesting on fifty percent (50%) of the outstanding and unvested equity awards held by Dr. Fisher that were subject to time-based vesting as of the Fisher Separation Date, which were deemed fully vested and exercisable as of the Fisher Separation Date; and (3) reimbursement of COBRA healthcare premium costs for the same level of coverage Dr. Fisher had during his employment with the Company, until the earliest of (i) twelve months from November 27, 2023, (ii) the date Dr. Fisher becomes eligible for substantially equivalent healthcare coverage through another source, or (iii) the expiration of Dr. Fisher’s eligibility for the continuation coverage. Further, and pursuant to the Separation Agreement, Dr. Fisher provided the Company with a general release of all claims, effective November 27, 2023.
Separation Agreement with Former COO
Guy Cipriani’s termination as the Company’s Chief Operating Officer effective October 2024 (the “Cipriani Separation Date”), in accordance with the terms of his Executive Employment Agreement with the Company, dated as of October 30, 2020 (the “Cipriani Employment Agreement”), and pursuant to Mr. Cipriani’s Separation Agreement with the Company, effective as of October 3, 2024 (the “Cipriani Separation Agreement”), the Company will provide Mr. Cipriani with (1) cash severance equivalent to twelve months of Mr. Cipriani’s base salary in effect as of the Separation Date, subject to standard payroll deductions and withholdings, payable over the Company’s regular payroll schedule over the twelve months following the Separation Date and (2) reimbursement of COBRA healthcare premium costs for the same level of coverage Mr. Cipriani had during his employment with the Company, until the earliest of (i) twelve months from October 3, 2024, (ii) the date Mr. Cipriani becomes eligible for substantially equivalent healthcare coverage through another source, or (iii) the expiration of Mr. Cipriani’s eligibility for the continuation coverage. Further, and pursuant to the Separation Agreement, Mr. Cipriani provided the Company with a general release of all claims, effective October 3, 2024.
Separation Agreement with Former CSO
Lee Arnold’s termination as the Company’s Chief Science Officer effective July 1, 2024 (the “Arnold Separation Date”), in accordance with the terms of his Executive Employment Agreement with the Company, dated as of February 1, 2023 (“Arnold Employment Agreement”), and pursuant to Mr. Arnold’s Separation Agreement with the Company, effective as of July 1, 2024 (the “Arnold Separation Agreement”), the Company will provide Mr. Arnold with (1) cash severance equivalent to twelve months of Mr. Arnold’s base salary in effect as of the Arnold Separation Date, subject to standard payroll deductions and withholdings, payable over the Company’s regular payroll schedule over the twelve months following the Separation Date and (2) reimbursement of COBRA healthcare premium costs for the same level of coverage Mr. Arnold had during his employment with the Company, until the earliest of (i) twelve months from July 1, 2024, (ii) the date Mr. Arnold becomes eligible for substantially equivalent healthcare coverage through another source, or (iii) the expiration of Mr. Arnold’s eligibility for the continuation coverage. Further, and pursuant to the Arnold Separation Agreement, Mr. Arnold provided the Company with a general release of all claims, effective July 1, 2024.
| 77 |
Employment Arrangements
We currently have written employment agreements with our executive officers. For information about our employment agreements with our named executive officers, refer to “Executive and Director Compensation — Employment Contracts.”
Equity Awards Granted to Executive Officers and Directors
We have granted stock options and RSUs to our executive officers and directors. For information about our grants of stock option awards and RSUs to our named executive officers and our directors, refer to “Executive and Director Compensation — Outstanding Equity Awards at 2024 Fiscal Year-End,” “Executive and Director Compensation — Director Compensation for 2024 Fiscal Year” and “Executive and Director Compensation — Non-Employee Director Compensation Policy.”
Indemnification Agreements
We have entered into and intend to continue to enter into indemnification agreements with each of our directors and our officers. The indemnification agreements, our Articles of Incorporation, as amended, and our Amended and Restated Bylaws require us to indemnify our directors and officers to the fullest extent permitted by Nevada law.
Policies and Procedures for Transactions with Related Persons
We maintain a written policy that our executive officers, directors, nominees for election as a director, beneficial owners of more than 5% of any class of our common stock and any members of the immediate family or affiliate of any of the foregoing persons are not permitted to enter into a related person transaction with us without the approval or ratification of the Audit Committee. Any request for us to enter into a transaction with an executive officer, director, nominee for election as a director, beneficial owner of more than 5% of any class of our common stock, or any member of the immediate family or affiliate of any of the foregoing persons, in which the amount involved exceeds $120,000 and such person would have a direct or indirect interest, must be presented to the Audit Committee for review, consideration and approval. In approving or rejecting any such proposal, the Audit Committee is to consider the material facts of the transaction, including whether the transaction is on terms no less favorable than terms generally available to an unaffiliated third party under the same or similar circumstances and the extent of the related person’s interest in the transaction.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The following table presents fees for professional services billed by Haskell & White and Baker Tilly during the fiscal years ended March 31, 2025 and 2024:
| Fiscal Year 2025 |
Fiscal Year 2024 |
|||||||
| Audit Fees(1) | $ | 237,300 | $ | 448,810 | ||||
| Tax Fees(2) | 17,500 | 40,386 | ||||||
| Total Fees | $ | 254,800 | $ | 489,196 | ||||
| (1) | Audit fees include fees for professional services rendered in connection with the audit of our annual financial statements for fiscal years 2025 and 2024 and for reviews of our quarterly financial statements and those services normally provided in connection with statutory or regulatory filings or engagements including comfort letters, consents and other services related to SEC matters. | |
| (2) | Tax Fees include the aggregate fees billed during fiscal year 2025 for professional services for preparation of income tax returns. |
Policy on Audit Committee Pre-approval of Audit and Permissible Non-audit Services of Independent Auditor
Our Audit Committee is responsible for pre-approving all audit, audit-related, tax and other permitted non-audit services to be performed for us by our independent auditor. The Audit Committee approved all of the services for which Baker Tilly billed us as set forth in the above table.
| 78 |
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
The following documents are filed as part of this Annual Report:
(a)(1) Financial Statements.
The response to this portion of Item 15 is set forth under Part II, Item 8 above.
(a)(2) Financial Statement Schedules.
All schedules have been omitted because they are not required or because the required information is given in the Financial Statements or Notes thereto set forth under Item 8 above.
(a)(3) Exhibits required by Item 601 of Regulation S-K.
| 79 |
| 80 |
| 81 |
| Incorporated by Reference | ||||||||||||
|
Exhibit Number |
Exhibit Description | Form | SEC File No. | Exhibit No. |
Date | Filed Herewith | ||||||
| 97.1 | Incentive Compensation Recoupment Policy. | X | ||||||||||
| 101.INS | Inline XBRL Instance Document (the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document) | X | ||||||||||
| 101.SCH | Inline XBRL Taxonomy Extension Schema Document | X | ||||||||||
| 101.CAL | Inline XBRL Taxonomy Extension Calculation Linkbase Document | X | ||||||||||
| 101.DEF | Inline XBRL Taxonomy Extension Definition Linkbase Document | X | ||||||||||
| 101.LAB | Inline XBRL Taxonomy Extension Label Linkbase Document | X | ||||||||||
| 101.PRE | Inline XBRL Taxonomy Extension Presentation Linkbase Document | X | ||||||||||
| 104 | Cover Page Interactive Data File (formatted in IXBRL, and included in exhibit 101) | X | ||||||||||
__________________
| ++ | Indicates management contract or compensatory plan. | |
| * | The information in Exhibit 32.1 shall not be deemed “filed” for purposes of Section 18 of the Exchange Act, or otherwise subject to the liabilities of that section, nor shall it be deemed incorporated by reference in any filing under the Securities Act or the Exchange Act (including this Annual Report), unless the Registrant specifically incorporates the foregoing information into those documents by reference. |
ITEM 16. FORM 10-K SUMMARY
None.
| 82 |
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, on the 26th day of June, 2025.
| By: | /s/ JAMES B. FRAKES | ||
|
JAMES B. FRAKES CHIEF EXECUTIVE OFFICER CHIEF FINANCIAL OFFICER. |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints James B. Frakes his or her true and lawful attorney-in-fact and agent, with full power of substitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agent full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agent or his substitutes or substitute, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
| /s/ JAMES B. FRAKES | Chief Executive Officer and Chief Financial Officer, | June 26, 2025 | ||
| James B. Frakes | Principal Executive Officer, Principal Financial and Accounting Officer and Director | |||
| /s/ EDWARD G. BROENNIMAN | Chairman and Director | June 26, 2025 | ||
| Edward G. Broenniman | ||||
| /s/ CHETAN S. SHAH | Director | June 26, 2025 | ||
| Chetan S. Shah, M.D. | ||||
| /s/ ANGELA ROSSETTI | Director | June 26, 2025 | ||
| Angela Rossetti | ||||
| /s/ NICOLAS GIKAKIS | Director | June 26, 2025 | ||
| Nicolas Gikakis | ||||
| 83 |
| Page | |
| Consolidated Financial Statements | |
|
Report of Independent Registered Public Accounting Firm (Haskell & White LLP, San Diego, CA PCAOB ID No |
F-2 |
|
Report of Independent Registered Public Accounting Firm (Baker Tilly,
San Diego, CA PCAOB ID No
|
F-4 |
| Consolidated Balance Sheets as of March 31, 2025 and 2024 | F-5 |
| Consolidated Statements of Operations and Comprehensive Loss for the Years Ended March 31, 2025 and 2024 | F-6 |
| Consolidated Statements of Equity for the Years Ended March 31, 2025 and 2024 | F-7 |
| Consolidated Statements of Cash Flows for the Years Ended March 31, 2025 and 2024 | F-8 |
| Notes to Consolidated Financial Statements | F-9 |
| F-1 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
Aethlon Medical, Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheet of Aethlon Medical, Inc. (the “Company”) as of March 31, 2025, and the related consolidated statements of operations and comprehensive loss, equity, and cash flows for the year then ended, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the consolidated financial position of the Company as of March 31, 2025, and the consolidated results of its operations and its cash flows for the year then ended, in conformity with accounting principles generally accepted in the United States of America.
The consolidated financial statements of the Company as of and for the year ended March 31, 2024 (the “fiscal 2024 consolidated financial statements”), before the effects of the retroactive adjustments described in Note 4 with respect to the one-for-eight reverse stock split effective as of the close of business on June 6, 2025 with an effective trading date of June 9, 2025, and the comparative disclosures related to the adoption of updated segment reporting standards as discussed in Note 9, were audited by other auditors whose report dated June 27, 2024, expressed an unqualified opinion, with an explanatory paragraph expressing substantial doubt about the Company’s ability to continue as a going concern on those statements. We also audited the adjustments described in Note 1 that were applied retroactively to the fiscal 2024 consolidated financial statements to reflect the June 9, 2025 one-for-eight reverse stock split, as described in in Note 4 and with respect to segment reporting in Note 9, and the related disclosures therein. In our opinion, such adjustments and related disclosures are appropriate and have been properly applied. We were not engaged to audit, review, or apply any procedures to the fiscal 2024 consolidated financial statements of the Company other than with respect to the adjustment and disclosures referred to herein and, accordingly, we do not express an opinion or any other form of assurance on the fiscal 2024 consolidated financial statements taken as a whole.
Substantial Doubt About the Company’s Ability to Continue as a Going Concern
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has recurring losses from operations, an accumulated deficit, expects to incur losses for the foreseeable future and requires additional working capital to achieve its operating plans. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1 to the consolidated financial statements. The consolidated financial statements do not include any adjustments to reflect the possible future effects on the recoverability and classification of assets or the amounts and classification of liabilities that may result from the outcome of this uncertainty.
| F-2 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM (Continued)
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audit. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (“PCAOB”) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audit provides a reasonable basis for our opinion.
Critical Audit Matters
Critical audit matters are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. We determined that there are no critical audit matters.
/s/
HASKELL & WHITE LLP
We have served as the Company’s auditor since 2024.
June 26, 2025
| F-3 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the shareholders and the board of directors of Aethlon Medical, Inc.:
Opinion on the Financial Statements
We have audited, before the effects of the adjustments to retrospectively apply the changes in presentation of the Company’s segment disclosure described in Notes 1 and 9 and the effects of the 1-for-8 reverse stock split described in Note 4, the accompanying consolidated balance sheet of Aethlon Medical, Inc. and its subsidiary (the "Company") as of March 31, 2024, the related consolidated statements of operations and comprehensive loss, equity, and cash flows for the year then ended, and the related notes (collectively referred to as the "consolidated financial statements"). In our opinion, the consolidated financial statements, before the effects of the adjustments to retrospectively apply the changes in presentation of the Company’s segment disclosure described in Notes 1 and 9 and the effects of the 1-for-8 reverse stock split described in Note 4, present fairly, in all material respects, the financial position of the Company as of March 31, 2024, and the results of their operations and their cash flows for the year then ended, in conformity with accounting principles generally accepted in the United States of America.
We were not engaged to audit, review, or apply any procedures to the adjustments to retrospectively apply the changes in segment presentation described in Notes 1 and 9 and reverse stock split in Note 4, and accordingly, we do not express an opinion or any other form of assurance about whether such adjustments are appropriate and have been properly applied. The adjustments were audited by other auditors.
Going Concern Uncertainty
The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 1 of the consolidated financial statements, the Company has recurring losses from operations, an accumulated deficit, expects to incur losses for the foreseeable future and requires additional working capital. These are the reasons that raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not contain any adjustments that might result from the outcome of this uncertainty.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company's consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) ("PCAOB") and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audit we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audit included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audit also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audit provides a reasonable basis for our opinion.
/s/
We served as the Company's auditor from 2001 to 2024.
June 27, 2024
| F-4 |
AETHLON MEDICAL, INC. AND SUBSIDIARY
CONSOLIDATED BALANCE SHEETS
| March 31, | ||||||||
| 2025 | 2024 | |||||||
| ASSETS | ||||||||
| CURRENT ASSETS | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Deferred offering costs | ||||||||
| Prepaid expenses and other current assets | ||||||||
| TOTAL CURRENT ASSETS | ||||||||
| Property and equipment, net | ||||||||
| Operating lease right-of-use asset | ||||||||
| Patents, net | ||||||||
| Restricted cash | ||||||||
| Deposits | ||||||||
| TOTAL ASSETS | $ | $ | ||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY | ||||||||
| CURRENT LIABILITIES | ||||||||
| Accounts payable | $ | $ | ||||||
| Due to related parties | ||||||||
| Operating lease liability, current portion | ||||||||
| Other current liabilities | ||||||||
| TOTAL CURRENT LIABILITIES | ||||||||
| Operating lease liability, less current portion | ||||||||
| TOTAL LIABILITIES | ||||||||
| COMMITMENTS AND CONTINGENCIES (Note 8) | ||||||||
| STOCKHOLDERS’ EQUITY | ||||||||
| Common stock, $ par value, shares authorized at March 31, 2025 and 2024; and shares issued and and outstanding at March 31, 2025 and 2024, respectively | ||||||||
| Additional paid-in capital | ||||||||
| Accumulated other comprehensive loss | ( |
) | ( |
) | ||||
| Accumulated deficit | ( |
) | ( |
) | ||||
| TOTAL STOCKHOLDERS’ EQUITY | ||||||||
| TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY | $ | $ | ||||||
See accompanying notes to the consolidated financial statements.
| F-5 |
AETHLON MEDICAL, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
| Years Ended March 31, | ||||||||
| 2025 | 2024 | |||||||
| OPERATING COSTS AND EXPENSES | ||||||||
| Professional fees | ||||||||
| Payroll and related expenses | ||||||||
| General and administrative | ||||||||
| Total operating expenses | ||||||||
| OPERATING LOSS | ( |
) | ( |
) | ||||
| OTHER EXPENSE (INCOME), NET | ||||||||
| Interest income | ( |
) | ( |
) | ||||
| Other income | ( |
) | ||||||
| Interest expense | ||||||||
| Other expense | ||||||||
| Total other expense (income), net | ( |
) | ||||||
| NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS | ( |
) | ( |
) | ||||
| Basic and diluted net loss per share attributable to common stockholders | $ | ) | $ | ) | ||||
| Weighted average number of common shares outstanding - basic and diluted | ||||||||
| NET LOSS ATTRIBUTABLE TO COMMON STOCKHOLDERS | ( |
) | ( |
) | ||||
| OTHER COMPREHENSIVE LOSS | ( |
) | ( |
) | ||||
| COMPREHENSIVE LOSS | $ | ( |
) | $ | ( |
) | ||
See accompanying notes to the consolidated financial statements.
| F-6 |
AETHLON MEDICAL, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF EQUITY
FOR THE YEARS ENDED MARCH 31, 2025 AND 2024
| COMMON STOCK | ADDITIONAL PAID IN |
ACCUMULATED | ACCUMULATED COMPREHENSIVE | TOTAL | ||||||||||||||||||||
| SHARES | AMOUNT | CAPITAL | DEFICIT | LOSS | EQUITY | |||||||||||||||||||
| BALANCE - MARCH 31, 2023 | $ | $ | $ | ( |
) | $ | ( |
) | $ | |||||||||||||||
| Issuances of common stock for cash under at the market program, net | ||||||||||||||||||||||||
| Rounding for reverse split | ||||||||||||||||||||||||
| Issuance of common shares upon vesting of restricted stock units and net stock option exercises | ( |
) | ( |
) | ||||||||||||||||||||
| Reversal of accrued commission liability | – | |||||||||||||||||||||||
| Stock-based compensation expense | – | |||||||||||||||||||||||
| Net loss | – | ( |
) | ( |
) | |||||||||||||||||||
| Other comprehensive loss | – | ( |
) | ( |
) | |||||||||||||||||||
| BALANCE – MARCH 31, 2024 | $ | $ | $ | ( |
) | $ | ( |
) | $ | |||||||||||||||
| Issuances of common stock for public offering | ||||||||||||||||||||||||
| Issuances of common stock for Class A and Class B warrant exercises, net | ||||||||||||||||||||||||
| Issuance of common shares upon vesting of restricted stock units and net stock option exercises | ( |
) | ( |
) | ||||||||||||||||||||
| Stock-based compensation expense | – | |||||||||||||||||||||||
| Warrant inducement expense, net | – | |||||||||||||||||||||||
| Net loss | – | ( |
) | ( |
) | |||||||||||||||||||
| Other comprehensive loss | – | ( |
) | ( |
) | |||||||||||||||||||
| BALANCE – MARCH 31, 2025 | $ | $ | $ | ( |
) | $ | ( |
) | $ | |||||||||||||||
See accompanying notes to the consolidated financial statements.
| F-7 |
AETHLON MEDICAL, INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CASH FLOWS
FOR THE YEARS ENDED MARCH 31, 2025 AND 2024
| Years Ended March 31, | ||||||||
| 2025 | 2024 | |||||||
| Cash flows from operating activities: | ||||||||
| Net loss | $ | ( |
) | $ | ( |
) | ||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||
| Depreciation and amortization | ||||||||
| Stock-based compensation | ||||||||
| Loss on disposal of property, plant and equipment | ||||||||
| Non-cash lease expense | ||||||||
| Warrant Inducement Expense | ||||||||
| Changes in operating assets and liabilities: | ||||||||
| Prepaid expenses and other current assets | ( |
) | ||||||
| Accounts payable and other current liabilities | ( |
) | ||||||
| Due to related parties | ||||||||
| Net cash used in operating activities | ( |
) | ( |
) | ||||
| Cash flows from investing activities: | ||||||||
| Purchases of property and equipment | ( |
) | ||||||
| Cash used in investing activities | ( |
) | ||||||
| Cash flows from financing activities: | ||||||||
| Tax withholding payments or tax equivalent payments for net share settlement of restricted stock units | ( |
) | ( |
) | ||||
| Net proceeds from the issuance of common stock | ||||||||
| Proceeds from exercise of warrants – standard terms | ||||||||
| Proceeds from exercise of warrants – induced terms | ||||||||
| Commission paid related to warrant inducement | ( |
) | ||||||
| Legal fees paid related to warrant inducement | ( |
) | ||||||
| Net cash provided by financing activities | ||||||||
| Effect of Exchange Rate on Changes on Cash | ( |
) | ||||||
| Net increase (decrease) in cash and cash equivalents and restricted cash | ( |
) | ||||||
| Cash and cash equivalents restricted cash at beginning of year | ||||||||
| Cash and cash equivalents and restricted cash at end of year | $ | $ | ||||||
| Supplemental information of non-cash investing and financing activities: | ||||||||
| Issuance of shares under vested restricted stock units, net stock option exercises and unvested share issuance for services | $ | $ | ||||||
| Disposal of fully depreciated property | $ | $ | ||||||
| Reversal of accrued commission liability (see Note 6) | $ | $ | ||||||
| Deferred offering costs not yet paid | $ | $ | ||||||
| Reconciliation of cash, cash equivalents and restricted cash to the consolidated balance sheets: | ||||||||
| Cash and cash equivalents | $ | $ | ||||||
| Restricted cash | ||||||||
| Cash and restricted cash | $ | $ | ||||||
See accompanying notes to the consolidated financial statements.
| F-8 |
Aethlon Medical, Inc. and Subsidiary
Notes to Consolidated Financial Statements
1. ORGANIZATION, LIQUIDITY AND SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
ORGANIZATION
Aethlon Medical, Inc. (“Aethlon,” the “Company,” “we” or “us”) is a medical therapeutic company focused on developing the Hemopurifier, a clinical-stage immunotherapeutic device designed to combat cancer and life-threatening viral infections and with additional potential applications e in organ transplantation and other areas of significant unmet needs. In human studies, 164 sessions with 38 patients, the Hemopurifier was safely utilized and demonstrated the potential to remove life-threatening viruses. In pre-clinical studies, the Hemopurifier has demonstrated the potential to remove harmful exosomes and exosomal particles from biological fluids, utilizing its proprietary lectin-based technology. This action has potential applications in cancer, where exosomes and exosomal particles may promote immune suppression and metastasis, and in life-threatening infectious diseases. The U.S. Food and Drug Administration (“FDA”) has designated the Hemopurifier as a “Breakthrough Device” for two independent indications:
| · | the treatment of individuals with advanced or metastatic cancer who are either unresponsive to or intolerant of standard of care therapy, and with cancer types in which exosomes or exosomal particles have been shown to participate in the development or severity of the disease; and | |
| · | the treatment of life-threatening viruses that are not addressed with approved therapies. |
Oncology
We believe that the Hemopurifier may be a substantial advancement in the treatment of patients with advanced and metastatic cancer through its design to bind to and remove harmful remove harmful extracellular vesicles particles that promote the growth and spread of tumors. In October 2022, we formed a wholly-owned subsidiary in Australia to initially conduct oncology-related clinical research, then seek regulatory approval and commercialize our Hemopurifier in Australia.
We completed an in vitro binding study of extracellular vesicles from cancer patient samples, to provide pre-clinical evidence to support our trial design and translational endpoints. Our study indicated positive results from this study, providing evidence that our Hemopurifier removes extracellular vesicles, or EVs, from plasma. This translational study provides pre-clinical evidence to support our phase 1 safety, feasibility and dose-finding clinical trials of our Hemopurifier in patients with solid tumors who have stable or progressive disease during anti-PD-1 monotherapy treatment, such as Keytruda® or Opdivo®.
We have launched in an Australia safety, feasibility and dose-finding clinical trials of the Hemopurifier in cancer patients with solid tumors who have stable or progressive disease during anti-PD-1 monotherapy treatment, such as Keytruda® (pembrolizumab) or Opdivo® (nivolumab). The primary endpoint of the approximately nine to 18-patient, is safety. Exploratory analyses will be conducted to explore the number of HP treatments required to produce sustained reductions of EVs as well as improve anti-tumor T cell activity. We plan to open a similarly designed trial in India.
The following three hospitals in Australia have received ethics committee approval, have gone through training on our device and are open for patient enrollment: Royal Adelaide Hospital in Adelaide, Australia and Pindara Private Hospital in the Gold Coast section of Australia and GenesisCare North Shore Hospital in Sydney, Australia. As of 16JUN2025 we have treated three participants in the first of the three treatment cohorts. Once these patients have completed the pre-specified 7-day safety follow-up period, the data will be presented to an independent Data Safety Monitoring Board (DSMB). The DSMB will provide a recommendation to Aethlon senior leadership on advancing to the next cohort where participants will receive 2 HP treatments during the one week treatment period.
The Company continues to pursue approval of a similar clinical trial in India. HREC approval has previously been obtained at Medanta Medicity Hospital. Following this a meeting with Subject Expert Committee (SEC) of the India Regulatory Agency CDSCO was held 5JUN2025. We are awaiting the formal approval letter of the CDSCO. The clinical trial at Medanta can commence following a Site Initiation Visit (SIV) by the company’s India CRO, Qualtran.
| F-9 |
Life-Threatening Viral Infections
The Company also believes that the Hemopurifier can be part of the broad-spectrum treatment of life-threatening highly glycosylated, or carbohydrate coated, viruses that are not addressed with an already approved treatment. In small-scale or early feasibility human studies, the Hemopurifier has been used in the past to treat individuals infected with human immunodeficiency virus, or HIV, hepatitis-C and Ebola.
Additionally, in vitro, the Hemopurifier has been demonstrated to capture Ebola, Marburg virus, Zika, Lassa, MERS-CoV, Cytomegalovirus, Epstein-Barr, Herpes simplex, Chikungunya, Dengue, West Nile, H1N1 swine flu, H5N1 bird flu, and the reconstructed 1918 Spanish flu virus. In several cases, these studies were conducted in collaboration with leading government or non-government research institutes.
The Hemopurifier has previously been studied under FDA and international regulatory frameworks for the treatment of severe SARS-CoV-2 infection. While we terminated our U.S. and India-based COVID-19 studies due to low ICU patient volume and shifting priorities, these programs demonstrated real-world use of the Hemopurifier in critically ill patients. We maintain an open IDE for viral indications to preserve optionality for future outbreaks or emergent pathogens.
We have sufficient inventory of Hemopurifiers to support our ongoing oncology trial in Australia as well as any near-term expansion of that study or potential trial activity in India. While we have received FDA approval to begin manufacturing at our San Diego facility under our IDE supplement, we are still awaiting FDA approval of a separate supplement to qualify an additional supplier of a key Hemopurifier component. We continue to work with the FDA on this process.
Pre-Clinical Exploration of Additional Clinical Uses for the Hemopurifier
The Aethlon R&D laboratory continues to explore potential new indications for the Hemopurifier. We have published in the peer-reviewed journal Transplant Immunology the ability of the device to remove extracellular vesicles and their microRNA cargo from acellular perfusates of discarded kidneys that had undergone normothermic machine perfusion.
On May 12, 2025, the results of our pre-clinical ex vivo study entitled “Ex Vivo Removal of CD41 positive platelet microparticles from Plasma by a Medical Device containing a Galanthus nivalis agglutinin (GNA) affinity resin” were published in the pre-print vehicle bioRxiv. This manuscript has been submitted to a peer-reviewed publication for review.
Platelet -derived extracellular vesicles (PD-EVs) are the most numerous EV population in the body and are released by platelets in response to a variety of stimuli. The cargo contained within these EVs have been noted to take part in damage to blood vessels, activation of immune cells and spread of tumor cells. Excessive levels of PD-EVs have been implicated in a myriad of diseases including cancer, lupus, systemic sclerosis, multiple sclerosis, Alzheimer’s disease, sepsis, acute and Long COVID.
We hypothesized that the Aethlon Hemopurifier which contains a propriety GNA affinity resin would remove platelet derived EVs from plasma. In this experiment two hundred milliliters on donated healthy human plasma were circulated over the Aethlon Hemoupurifier (HP) to simulate a clinical HP session. The study results showed a 98.5% removal of platelet -derived EVs at a timepoint equivalent to a 4-hour HP treatment. The results of this study support the current Australian Clinical Trial in Oncology as well as open the investigation of the Hemopurifier in many indications.
Extracellular vesicles have been implicated in the pathogenesis of Long COVID.As we had previously demonstrated removal of extracellular vesicles by the Hemopurifier in a patient with severe acute COVID-19 infection, we hypothesized that patients with Long COVID would have extracellular vesicles with the mannose sugar on their surface that would bind to the affinity resin in our device. We partnered with investigators at the Univ of California San Francisco Medical Center Long COVID clinic to obtain samples from participants with Long COVID as well as controls that had had COVID -10 infection but had recovered. The data to be presented will review the binding of larger and smaller extracellular vesicles to the GNA lectin and the lectin affinity resin, respectively. We believe the data from this pre-clinical study calls for additional study of the Hemopurifier and look forward to receiving feedback from the Long COVID scientific community at the Keystone Symposium.
| F-10 |
Successful outcomes of human trials will also be required by the regulatory agencies of certain foreign countries where we plan to market and sell the Hemopurifier. Some of our patents may expire before FDA approval or approval in a foreign country, if any, is obtained. However, we believe that certain patent applications and/or other patents issued to us more recently will help protect the proprietary nature of our Hemopurifier treatment technology.
We believe that the Hemopurifier may be a substantial advancement in the treatment of patients with advanced and metastatic cancer
In addition to the foregoing, we are monitoring closely the impact of inflation, recent bank failures and the war between Russia and Ukraine and the military conflicts in Israel and the surrounding areas, as well as related political and economic responses and counter-responses by various global factors on our business. Given the level of uncertainty regarding the duration and impact of these events on capital markets and the U.S. economy, we are unable to assess the impact on our timelines and future access to capital. The full extent to which inflation, recent bank failures and the ongoing military conflicts will impact our business, results of operations, financial condition, clinical trials and preclinical research will depend on future developments, as well as the economic impact on national and international markets that are highly uncertain.
LIQUIDITY AND GOING CONCERN
The Company has incurred losses since inception
in devoting substantially all of its efforts toward research and development and has an accumulated deficit of $
The Company’s ability to execute its current operating plan depends on its ability to reduce expenses and obtain additional funding via the sale of equity, or other sources of capital. The Company plans to continue actively pursuing financing alternatives, however, there can be no assurance that it will obtain the necessary funding, raising substantial doubt about the Company’s ability to continue as a going concern within one year of the date these financial statements are issued. The accompanying financial statements do not include any adjustments that might result from the outcome of this uncertainty.
PRINCIPLES OF CONSOLIDATION
The accompanying consolidated financial statements include the accounts of Aethlon Medical, Inc. and its wholly owned subsidiary, Aethlon Medical Australia Pty Ltd.. Operations in our Australian subsidiary is recorded in their functional currency. The results of operations for our Australian subsidiary are translated from functional currency into U.S. dollars. Expenses originally incurred in U.S. dollars are translated using the exchange rate on the transaction date. For expenses in the subsidiary’s functional currency, we use the average exchange rate for the period, as it is not practical to determine the exact rate for each transaction date. Assets and liabilities are translated using the period end exchange rates. The U.S dollar effects that arise from translating the net assets of are recorded in other comprehensive income (loss). All significant inter-company transactions and balances have been eliminated in consolidation. The consolidated financial statements contain all normal recurring accruals and adjustments that, in the opinion of management, are necessary to present fairly the consolidated financial statements as of and for the fiscal years ended March 31, 2025 and 2024, and the consolidated statement of cash flows for the fiscal years ended March 31, 2025 and 2024.
RISKS AND UNCERTAINTIES
We operate in an industry that is subject to intense competition, government regulation and rapid technological change. Our operations are subject to significant risk and uncertainties including financial, operational, technological, regulatory, and including the potential risk of business failure.
| F-11 |
USE OF ESTIMATES
We prepare our consolidated financial statements in conformity with accounting principles generally accepted in the United States of America, or GAAP, which requires us to make a number of estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements. Such estimates and assumptions affect the reported amounts of expenses during the reporting period. On an ongoing basis, we evaluate estimates and assumptions based upon historical experience and various other factors and circumstances. We believe our estimates and assumptions are reasonable in the circumstances; however, actual results may differ from these estimates under different future conditions.
We believe that the estimates and assumptions that are most important to the portrayal of our financial condition and results of operations, in that they require the most difficult, subjective or complex judgments, form the basis for the accounting policies deemed to be most critical to us.
CASH AND CASH EQUIVALENTS
Accounting standards define “cash and cash equivalents” as any short-term, highly liquid investment that is both readily convertible to known amounts of cash and so near their maturity that they present insignificant risk of changes in value because of changes in interest rates. For the purpose of financial statement presentation, we consider all highly liquid investment instruments with original maturities of three months or less when purchased, or any investment redeemable without penalty or loss of interest to be cash equivalents. Cash is carried at cost, which approximates fair value, and cash equivalents are carried at fair value.
As of March 31, 2025 and March 31, 2024 our cash and cash equivalents were comprised of the following instruments:
| For the year ended | ||||||||
| March 31, 2025 |
March 31, 2024 |
|||||||
| Cash in US bank checking account | $ | $ | ||||||
| Cash equivalents held in US Treasury bills | ||||||||
| Cash in Australian bank checking account | ||||||||
| Total cash and cash equivalents | $ | $ | ||||||
CONCENTRATIONS OF CREDIT RISKS
Cash is maintained at one US financial institution
in a checking account. Accounts at this institution are secured by the Federal Deposit Insurance Corporation up to $250,000. Our March
31, 2025 cash balances were approximately $
At March
31, 2025, we maintained cash equivalents of approximately $
Cash is maintained at one Australian financial institution in checking accounts. Accounts at this institution are secured by the Financial Claims Scheme for up to Australian $250,000. Our March 31, 2025 Australian cash balance was below that threshold.
RESTRICTED CASH
To comply with the terms
of our laboratory, office, and manufacturing space leases, we arranged for our former bank, First Republic Bank, to issue two standby
letters of credit (L/Cs) totaling $
| F-12 |
PROPERTY AND EQUIPMENT
Property and equipment are stated at cost. Depreciation is computed using the straight-line method over the estimated useful lives of the related assets, which range from two to five years. Repairs and maintenance are charged to expense as incurred while improvements are capitalized. Upon the sale or retirement of property and equipment, the accounts are relieved of the cost and the related accumulated depreciation with any gain or loss included in the consolidated statements of operations.
INCOME TAXES
Deferred tax assets and liabilities are recognized
for the future tax consequences attributable to the difference between the consolidated financial statements and their respective tax
basis. Deferred income taxes reflect the net tax effects of (a) temporary differences between the carrying amounts of assets and liabilities
for financial reporting purposes and the amounts reported for income tax purposes, and (b) tax credit carryforwards. We record a valuation
allowance for deferred tax assets when, based on our best estimate of taxable income (if any) in the foreseeable future, it is more likely
than not that some portion of the deferred tax assets may not be realized. Management has provided a full valuation allowance against
the Company’s net deferred tax asset. Tax positions taken or expected to be taken in the course of preparing tax returns are required
to be evaluated to determine whether the tax positions are more-likely-than-not to be sustained by the applicable tax authority. Tax positions
deemed to not meet a more-likely-than-not threshold would be recorded as tax expense in the current year. There were
LONG-LIVED ASSETS
Long-lived assets are reviewed for impairment
whenever events or changes in circumstances indicate that their carrying amounts may not be recoverable. If the cost basis of a long-lived
asset is greater than the projected future undiscounted net cash flows from such asset, an impairment loss is recognized. We believe
Basic loss per share is computed by dividing net loss available to common stockholders by the weighted average number of common shares outstanding during the period of computation. Diluted loss per share is computed similar to basic loss per share except that the denominator is increased to include the number of additional common shares that would have been outstanding if potential common shares had been issued, if such additional common shares were dilutive. Since we had net losses for all periods presented, basic and diluted loss per share are the same, and additional potential common shares have been excluded as their effect would be antidilutive.
As of March 31, 2025 and 2024, a total of and potential common shares, consisting of shares underlying outstanding stock options, restricted stock units, or RSUs, shares held in abeyance and warrants were excluded as their inclusion would be antidilutive.
REVENUE RECOGNITION
We did not recognize revenue in fiscal year ended March 31, 2025 or March 31, 2024.
Employee stock options and rights to purchase shares under stock participation plans are accounted for under the fair value method. Accordingly, share-based compensation is measured when all granting activities have been completed, generally the grant date, based on the fair value of the award. The exercise price of options is generally equal to the market price of the Company’s common stock (defined as the closing price as quoted on the Nasdaq Capital Market or OTCBB on the date of grant). Compensation cost recognized by the Company includes (a) compensation cost for all equity incentive awards granted prior to April 1, 2006, but not yet vested, based on the grant-date fair value estimated in accordance with the original provisions of the then current accounting standards, and (b) compensation cost for all equity incentive awards granted subsequent to March 31, 2006, based on the grant-date fair value estimated in accordance with the provisions of subsequent accounting standards. We use a Binomial Lattice option pricing model for estimating fair value of options granted (see Note 4).
| F-13 |
The following table summarizes share-based compensation expenses relating to shares and options granted and the effect on loss per common share during the years ended March 31, 2025 and 2024:
| Fiscal Years Ended | ||||||||
| March 31, 2025 | March 31, 2024 | |||||||
| Vesting of Stock Options and Restricted Stock Units | $ | $ | ||||||
| Total Stock-Based Compensation Expense | $ | $ | ||||||
| Weighted average number of common shares outstanding – basic and diluted | ||||||||
| Basic and diluted loss per common share | $ | ) | $ | ) | ||||
We record share-based compensation expenses for awards of stock options and RSUs under ASC 718, Share-based compensation, or ASC 718. For awards to non-employees for periods prior to the adoption of ASU 2018-07, Compensation-Stock Compensation: Improvements to Non-employee Share-Based Payment Accounting, on April 1, 2019, the Company had applied ASC 505-50, Equity – Equity-based payments to non-employees, or ASC 505-50. ASC 718 establishes guidance for the recognition of expenses arising from the issuance of share-based compensation awards at their fair value at the grant date.
We recognize share-based compensation expense related to stock options and stock appreciation rights granted to employees, directors and consultants based on the estimated fair value of the awards on the date of grant. We estimate the grant date fair value, and the resulting share-based compensation expense, for stock options that only have service vesting requirements or performance-based vesting requirements without market conditions using the binomial lattice option-pricing model. The grant date fair value of the share-based awards with service vesting requirements is generally recognized on a straight-line basis over the requisite service period, which is generally the vesting period of the respective awards. Determining the appropriate amount to expense for performance-based awards based on the achievement of stated goals requires judgment. The estimate of expense is revised periodically based on the probability of achieving the required performance targets and adjustments are made as appropriate. The cumulative impact of any revisions is reflected in the period of change. If any applicable financial performance goals are not met, no compensation cost is recognized and any previously recognized compensation cost is reversed. For performance-based awards with market conditions, we determine the fair value of awards as of the grant date using a Monte Carlo simulation model.
We review share-based compensation on a quarterly basis for changes to the estimate of expected award forfeitures based on actual forfeiture experience. The effect of adjusting the forfeiture rate for all expense amortization after March 31, 2007 is recognized in the period the forfeiture estimate is changed. The effect of forfeiture adjustments for the fiscal year ended March 31, 2025 was insignificant.
PATENTS
Patents include both foreign and domestic patents. We capitalize the cost of patents, some of which were acquired, and amortize such costs over the shorter of the remaining legal life or their estimated economic life, upon issuance of the patent. The unamortized costs of patents are subject to our review for impairment under our long-lived asset policy above.
STOCK PURCHASE WARRANTS
In the past we issued warrants for the purchase of shares of our common stock in connection with the issuance of common stock for cash. Warrants issued in connection with common stock for cash, if classified as equity, are considered issued in connection with equity transactions and the warrant fair value is recorded to additional paid-in-capital.
RESEARCH AND DEVELOPMENT EXPENSES
Our research and development costs are expensed
as incurred. We incurred approximately $
| F-14 |
OFF-BALANCE SHEET ARRANGEMENTS
We have not entered into any off-balance sheet arrangements that have or are reasonably likely to have a current or future material effect on our consolidated financial statements.
SIGNIFICANT RECENT ACCOUNTING PRONOUNCEMENTS
In fiscal year 2025, the Company adopted Accounting Standards Update (ASU) No. 2023-07, Segment Reporting (Topic 280): Improvements to Reportable Segment Disclosures. This ASU requires public entities to disclose significant segment expense categories that are regularly provided to the Chief Operating Decision Maker (CODM) and included in the measure of segment profit or loss.
The Company operates as a single reportable segment. The adoption of ASU 2023-07 did not impact the Company’s consolidated financial statements but resulted in enhanced footnote disclosures regarding significant segment expenses, as reflected in Note 9 – Segment Reporting.
In March 2024, the FASB issued Accounting Standards Update 2024-03, Income Statement—Reporting Comprehensive Income—Expense Disaggregation Disclosures (“ASU 2024-03”), which requires public business entities to provide enhanced annual and interim disclosures that disaggregate specified income statement expense categories. ASU 2024-03 is effective for annual periods beginning after December 15, 2026, and interim periods within fiscal years beginning after December 15, 2027. The Company is evaluating whether the adoption of this new standard will have a material impact on our disclosures.
In March 2024, the Financial Accounting Standards Board (FASB) issued Accounting Standards Update No. 2024-02, Codification Improvements—Amendments to Remove References to the Concepts Statements (“ASU 2024-02”). ASU 2024-02 eliminates various references to the FASB’s Concepts Statements from the FASB Accounting Standards Codification in order to clarify that the Codification represents the authoritative source of generally accepted accounting principles (GAAP) in the United States. The amendments do not alter existing accounting requirements. The guidance is effective for fiscal years, including interim periods within those fiscal years, beginning after December 15, 2024, and early adoption is permitted. This ASU is not expected to have a material impact on the Company’s consolidated financial statements or disclosures.
In December 2023, the FASB issued Accounting Standards Update 2023-09, Improvements to Income Tax Disclosures (“ASU 2023-09”), which requires enhanced annual disclosures for specific categories in the rate reconciliation and income taxes paid disaggregated by federal, state and foreign taxes. ASU 2023-09 is effective for public business entities for annual periods beginning after December 15, 2024. The Company is evaluating if the adoption of this new standard will have a material effect on our disclosures.
In June 2016, the FASB issued ASU No. 2016-13, Financial Instruments-Credit Losses (Topic 326), Measurement of Credit Losses on Financial Instruments. The adoption of ASU No. 2016-13 for smaller reporting companies that did not previously early adopt was January 1, 2023. The Company maintained US Treasury bills with maturities of less than three months and expects zero credit losses from these securities. As a result, the Company did not record an allowance for expected credit losses.
2. PROPERTY AND EQUIPMENT, NET
Property and equipment, net, consist of the following:
| March 31, 2025 | March 31, 2024 | |||||||
| Furniture and office equipment, at cost | $ | $ | ||||||
| Less: disposals | ( |
) | ||||||
| Leasehold improvements | ||||||||
| Gross property and equipment | ||||||||
| Less: accumulated depreciation | ( |
) | ( |
) | ||||
| Fixed assets, net | $ | $ | ||||||
| F-15 |
Depreciation expense for the fiscal years ended March 31, 2025 and
2024 was $
3. PATENTS, NET
Patents, net consist of the following:
| March 31, 2025 | March 31, 2024 | |||||||
| Issued patents | $ | $ | ||||||
| Accumulated amortization | ( |
) | ( |
) | ||||
| Patents, net | $ | $ | ||||||
Amortization expense for our capitalized issued
patents for each of the fiscal years ended March 31, 2025 and 2024 was $
4. EQUITY TRANSACTIONS
REVERSE STOCK SPLIT
Effective as of
the close of business on June 6, 2025 with an effective trading date of June 9, 2025, we
effected a
WARRANT INDUCEMENT
In March 2025, the Company
entered into a warrant inducement agreement resulting in the issuance of new warrants and the modification of existing
warrants. The fair value of the new warrants and the incremental fair value from the modification totaled $
ISSUANCES OF COMMON STOCK AND WARRANTS
Equity Transactions in the Fiscal Year Ended March 31, 2025.
March 2025 Warrant-Based Financing
On March 16, 2025, the
Company entered into an inducement offer to exercise existing Class A and Class B Warrants
(the “Agreement”) with a certain accredited and institutional holder (the “Holder”) of the Company’s outstanding
Class A and Class B Warrants issued on May 17, 2024 (the “Existing Warrants”).
| F-16 |
The closing took place
on March 17, 2025. Gross proceeds to the Company from the exercise of the Existing Warrants was $
As a result of the Holder exercising the Existing Warrants, the Company issued an aggregate of shares of its common stock. The shares underlying the Existing Warrants have all been registered on Form S-1 registration statement (Registration Number 333-278188).
The Company agreed to file a resale registration statement registering the shares underlying the Replacement Warrants (“Resale Registration Statement”) within ninety (90) days of the date of the Agreement and to use commercially reasonable best efforts to cause the Resale Registration Statement to be effective on or prior to the 150th calendar day after the date of the Agreement.
Subject to the terms of the Agreement, the Company will be required to pay certain liquidated damages if the shares underlying the New Warrants are not filed within the ninety (90) period, as more fully described in the Agreement.
The Company further agreed that until sixty (60) days after the closing date of the warrant exercise, it will not (other than in connection with limited enumerated exceptions) issue, enter into any agreement to issue or announce the issuance or proposed issuance of any shares of common stock or common stock equivalents or file any registration statement or any amendment or supplement (other than the registration statement registering the shares underlying the Replacement Warrants).
In connection with the
transactions contemplated in the Agreement, the Company agreed to pay its placement agent, Maxim Group, LLC (the “Agent”)
the following compensation, (i) a cash fee equal to 6.0% of the gross proceeds received by the Company in the transactions contemplated
by the Agreement, and (ii) legal fees and out-of-pocket expenses of $
May 2024 Public Offering
On May 17, 2024,
the Company closed a public offering pursuant to which it sold an aggregate of: (i) shares
of common stock and accompanying Class A warrants to purchase up to shares
of common stock and Class B warrants to purchase up to shares
of common stock, at a combined public offering price of $
per share and accompanying warrants; and (ii) in lieu of common stock, pre-funded warrants to purchase
All pre-funded
warrants issued in the offering were exercised in the quarter ended June 30, 2024. The Class A
and Class B warrants each have an exercise price of $
Maxim Group LLC (“Maxim”),
served as the exclusive placement agent in connection with the offering. We paid Maxim a cash fee of 6.5% of the aggregate gross proceeds
raised at the closing of the offering, and reimbursement of certain expenses and legal fees in the amount of $
The gross proceeds from
the offering, before deducting the placement agent’s fees and other offering expenses, were approximately $
The shares of Common Stock, the Class A and Class B warrants, the pre-funded warrants and the Placement Agent Warrants described above and the underlying shares of Common Stock were offered pursuant to a Registration Statement on Form S-1, as amended (File No. 333-278188) (the “Registration Statement”), which was declared effective by the Securities and Exchange Commission (the “SEC”) on May 15, 2024
| F-17 |
RSU Grants to Non-Employee Directors
In April 2024, the Compensation Committee of the Board, or Compensation Committee, approved, pursuant to the terms of the Company’s Amended and Restated Non-Employee Director Compensation Policy, or the Director Compensation Policy, the grant of the annual RSUs under the Director Compensation Policy to each of the three non-employee directors of the Company then serving on the Board of Directors of the Company, or Board. The Director Compensation Policy provides for a grant of stock options or $50,000 worth of RSUs at the beginning of each fiscal year for current non-employee directors then serving on the Board, and for a grant of stock options or $75,000 worth of RSUs for a newly elected non-employee director, with each RSU priced at the average for the closing prices for the five days preceding and including the date of grant, or $12.16 per share for the April 2024 RSU grants. As a result, in April 2024 the four eligible directors were each granted an RSU in the amount of shares under the 2020 Plan.
There were no vested RSUs outstanding as of March 31, 2025.
Equity Transactions in the Fiscal Year Ended March 31, 2024.
On March 24, 2022, we entered into an At The Market Offering Agreement, or the 2022 ATM Agreement, with H.C. Wainwright & Co., LLC, or Wainwright, which established an at-the-market equity program pursuant to which we may offer and sell shares of our common stock from time to time as set forth in the 2022 ATM Agreement.
The offering was registered under the Securities
Act of 1933, as amended, or the Securities Act, pursuant to our shelf registration statement on Form S-3 (Registration Statement No. 333-259909),
as previously filed with the Securities and Exchange Commission, or SEC, and declared effective on October 21, 2021. We filed a prospectus
supplement, dated March 24, 2022, with the SEC that provides for the sale of shares of our common stock, or the 2022 ATM Shares, having
an aggregate offering price of up to $
The underlying shelf registration statement on Form S-3 expired in October 2024. As a result, we were no longer eligible to sell shares under the 2022 ATM Agreement, which we subsequently terminated following the expiration
Under the 2022 ATM Agreement, Wainwright may sell the 2022 ATM Shares by any method permitted by law and deemed to be an “at the market offering” as defined in Rule 415 promulgated under the Securities Act, including sales made directly on the Nasdaq Capital Market, or on any other existing trading market for the 2022 ATM Shares. In addition, under the 2022 ATM Agreement, Wainwright may sell the 2022 ATM Shares in privately negotiated transactions with our consent and in block transactions. Under certain circumstances, we may instruct Wainwright not to sell the 2022 ATM Shares if the sales cannot be effected at or above the price designated by us from time to time.
We are not obligated to make any further sales of the 2022 ATM Shares under the 2022 ATM Agreement. The offering of the 2022 ATM Shares pursuant to the 2022 ATM Agreement will terminate upon the termination of the 2022 ATM Agreement by Wainwright or us, as permitted therein.
The 2022 ATM Agreement contains customary representations, warranties and agreements by us, and customary indemnification and contribution rights and obligations of the parties. We agreed to pay Wainwright a placement fee of up to 3.0% of the aggregate gross proceeds from each sale of the 2022 ATM Shares. We also agreed to reimburse Wainwright for certain specified expenses in connection with entering into the 2022 ATM Agreement.
In the fiscal year ended March 31, 2024, we raised
aggregate net proceeds of $
.
| F-18 |
RSU Grants to Non-Employee Directors
In April 2024, the Compensation Committee of the Board, or Compensation Committee, approved, pursuant to the terms of the Company’s Amended and Restated Non-Employee Director Compensation Policy, or the Director Compensation Policy, the grant of the annual RSUs under the Director Compensation Policy to each of the three non-employee directors of the Company then serving on the Board of Directors of the Company, or Board. The Director Compensation Policy provides for a grant of stock options or $50,000 worth of RSUs at the beginning of each fiscal year for current non-employee directors then serving on the Board, and for a grant of stock options or $75,000 worth of RSUs for a newly elected non-employee director, with each RSU priced at the average for the closing prices for the five days preceding and including the date of grant, or $34.40 per share for the April 2023 RSU grants. As a result, in April 2023 the three eligible directors were each granted an RSU in the amount of shares under the 2020 Plan.
Unvested RSUs covering shares of common stock were outstanding as of March 31, 2024.
WARRANTS:
During the twelve-months ended March 31, 2025, we issued warrants in connection with the May, 2024 public offering and in connection with a March 2025 warrant inducement. We did not issue any warrants during the fiscal year ended March 31, 2024.
A summary of the aggregate warrant activity for the years ended March 31, 2025 and 2024 is presented below:
| Fiscal Year Ended March 31, | ||||||||||||||||
| 2025 | 2024 | |||||||||||||||
| Warrants | Weighted Average Exercise Price |
Warrants | Weighted Average Exercise Price |
|||||||||||||
| Outstanding, beginning of year | $ | $ | ||||||||||||||
| Granted | $ | $ | N/A | |||||||||||||
| Exercised | ( |
) | $ | $ | N/A | |||||||||||
| Cancelled/Forfeited | ( |
) | $ | $ | N/A | |||||||||||
| Outstanding, end of year | $ | $ | ||||||||||||||
| Exercisable, end of year | $ | $ | ||||||||||||||
| Weighted average estimated fair value of warrants granted | $ | N/A | $ | N/A | ||||||||||||
The detail of the warrants outstanding and exercisable as of March 31, 2025 is as follows:
| Warrants Outstanding | Warrants Exercisable | |||||||||||||||||||
| Range of Exercise Prices |
Number Outstanding |
Weighted Average Remaining Life (Years) |
Weighted Average Exercise Price |
Number Outstanding |
Weighted Average Exercise Price |
|||||||||||||||
| $4.64 or Below | $ | $ | ||||||||||||||||||
The detail of the warrants outstanding and exercisable as of March 31, 2024 is as follows:
| Warrants Outstanding | Warrants Exercisable | |||||||||||||||||||
| Range of Exercise Prices |
Number Outstanding |
Weighted Average Remaining Life (Years) |
Weighted Average Exercise Price |
Number Outstanding |
Weighted Average Exercise Price |
|||||||||||||||
| $150.00 or Below | $ | $ | ||||||||||||||||||
| $200.00 - $220.00 | $ | $ | ||||||||||||||||||
| F-19 |
STOCK-BASED COMPENSATION:
2020 EQUITY INCENTIVE PLAN
The
2020 Equity Incentive Plan (the “2020 Plan”) was approved by our stockholders in September 2020 to provide stock-based incentives
to attract, retain, and motivate employees, directors, and consultants. The plan initially authorized
NON-EMPLOYEE DIRECTOR COMPENSATION POLICY
The Company maintains the Director Compensation Policy which provides for cash and equity compensation for persons serving as non-employee directors of the Company. Under this policy, each new non-employee director receives either stock options or a grant of RSUs upon appointment/election, as well as either an annual grant of stock options or of RSUs at the beginning of each fiscal year. The (i) stock options are subject to vesting and (ii) RSUs are subject to vesting and represent the right to be issued on a future date shares of our common stock upon vesting.
Please see above under the heading "Equity Transactions in the Fiscal Year Ended March 31, 2025—RSU Grants to Non-Employee Directors" for disclosure regarding equity awards under the Director Compensation Policy during the fiscal year ended March 31, 2025.
STOCK OPTION ACTIVITY
During the fiscal years ended March 31, 2025 and March 31, 2024, we did not issue stock option grants.
Options outstanding that were vested as of March 31, 2025 and options that are expected to vest subsequent to March 31, 2025 are as follows:
| Number of Shares |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Term in Years |
||||||||||
| Vested | $ | |||||||||||
| Expected to vest | $ | |||||||||||
| Total | ||||||||||||
The following is a summary of the stock options outstanding at March 31, 2025 and 2024 and the changes during the years then ended:
| Fiscal Year Ended March 31, | ||||||||||||||||
| 2025 | 2024 | |||||||||||||||
| Options | Weighted Average Exercise Price |
Options | Weighted Average Exercise Price |
|||||||||||||
| Outstanding, beginning of year | $ | $ | ||||||||||||||
| Granted | $ | $ | ||||||||||||||
| Cancelled/Forfeited | ( |
) | $ | ( |
) | $ | ||||||||||
| Outstanding, end of year | $ | $ | ||||||||||||||
| Exercisable, end of year | $ | $ | ||||||||||||||
| Weighted average estimated fair value of options granted | $ | N/A | $ | N/A | ||||||||||||
| F-20 |
Effective as of the close of business on June 6, 2025 with an effective trading date of June 9, 2025, the Company effected
a
There were stock option grants during the fiscal years ended March 31, 2025 or March 31, 2024. There were RSUs granted during the fiscal year ended March 31, 2025. The weighted average grant date fair value of RSUs granted during the fiscal year ended March 31, 2025 was $. There were stock option exercises during the fiscal years ended March 31, 2025 and 2024.
The table below summarizes nonvested stock options as of March 31, 2025 and changes during the year ended March 31, 2025.
| Shares | Weighted Average Grant Date Fair Value | |||||||
| Nonvested stock options at April 1, 2024 | $ | |||||||
| Vested | ( |
) | $ | |||||
| Forfeited | ( |
) | $ | |||||
| Nonvested stock options at March 31, 2025 | ||||||||
The detail of the options outstanding and exercisable as of March 31, 2025 is as follows:
| Options Outstanding | Options Exercisable | |||||||||||||||||||
| Exercise Prices | Number Outstanding |
Weighted Average Remaining Life (Years) |
Weighted Average Exercise Price |
Number Outstanding |
Weighted Average Exercise Price |
|||||||||||||||
| $102.40-112.8 | years | $ | $ | |||||||||||||||||
| $201.6 | years | $ | $ | |||||||||||||||||
| Total | ||||||||||||||||||||
We recorded stock-based compensation expense related to RSU issuances and to options granted totaling $ and $ for the fiscal years ended March 31, 2025 and 2024, respectively. These expenses were recorded as stock compensation included in payroll and related expenses in the accompanying consolidated statement of operations for the years ended March 31, 2025 and 2024.
The table below presents a summary of restricted stock unit activity as of March 31, 2025, and for the year then ended.
| Shares | ||||
| Nonvested RSUs at April 1, 2024 | ||||
| Granted | ||||
| Vested | ( |
) | ||
| Tax withholding payments or tax equivalent payments for net share settlement of restricted stock units | ( |
) | ||
| Nonvested RSUs at March 31, 2025 | ||||
Our total stock-based compensation for fiscal years ended March 31, 2025 and 2024 included the following:
| Fiscal Year Ended | ||||||||
| March 31, 2025 | March 31, 2024 | |||||||
| Vesting of restricted stock units | $ | $ | ||||||
| Vesting of stock options | ||||||||
| Total Stock-Based Compensation | $ | $ | ||||||
| F-21 |
We review share-based compensation on a quarterly basis for changes to the estimate of expected award forfeitures based on actual forfeiture experience. The cumulative effect of adjusting the forfeiture rate for all expense amortization is recognized in the period the forfeiture estimate is changed. The effect of forfeiture adjustments for the fiscal year ended March 31, 2025 was insignificant.
On March 31, 2025, our outstanding stock options had no intrinsic value since the closing price on that date of $ per share was below the weighted average exercise price of our outstanding stock options.
At March 31, 2025, there was $ of unrecognized compensation cost related to share-based payments, which is expected to be recognized over a weighted average period of years.
5. RELATED PARTY TRANSACTIONS
DUE TO RELATED PARTIES
For the fiscal year ended
March 31, 2025 we accrued unpaid fees of $
As a result of entering
into separation agreements with two former executives, we paid out accrued vacation totaling $
Separately, in connection
with a reduction in force in August 2024, we paid out an additional $
Amounts due to related parties were comprised of the following items:
| March 31, 2025 |
March 31, 2024 |
|||||||
| Accrued Board fees | $ | $ | ||||||
| Accrued vacation to all employees | ||||||||
| Accrued separation expenses | ||||||||
| Total due to related parties | $ | $ | ||||||
6. OTHER CURRENT LIABILITIES
Other current liabilities were comprised of the following items:
| March 31, 2025 | March 31, 2024 | |||||||
| D&O insurance premium financing (See Note 8) | $ | |||||||
| Accrued professional fees | $ | |||||||
| Accrued resale registration | ||||||||
| Total other current liabilities | $ | $ | ||||||
During 2017 through 2020, the Company incorrectly recorded accrued commission liability of approximately $404,000. The Company reversed accrued commission liability of approximately $404,000 during the year ended March 31, 2024.
| F-22 |
7. INCOME TAXES
For the years ended March 31, 2025 and 2024, we had no income tax expense due to our net operating losses and 100% deferred tax asset valuation allowance.
At March 31, 2025 and 2024, we had net deferred tax assets as detailed below. These deferred tax assets are primarily composed of capitalized research and development costs and tax net operating loss carryforwards. Due to uncertainties surrounding our ability to generate future taxable income to realize these assets, a 100% valuation allowance has been established to offset the net deferred tax assets.
Significant components of our net deferred tax assets at March 31, 2025 and 2024 are shown below:
| YEAR ENDED MARCH 31, | ||||||||
| 2025 | 2024 | |||||||
| Deferred tax assets: | ||||||||
| Research and development credit carryforwards | $ | $ | ||||||
| Capitalized research and development costs | ||||||||
| Net operating loss carryforwards(1) | ||||||||
| Stock compensation | ||||||||
| Total deferred tax assets | ||||||||
| Total deferred tax liabilities | ||||||||
| Net deferred tax assets | ||||||||
| Valuation allowance for deferred tax assets | ( |
) | ( |
) | ||||
| Net deferred tax assets | $ | $ | ||||||
______________
| (1) |
At March 31, 2025, we had tax net operating loss
carryforwards for federal and state purposes approximating $
The provision for income taxes on earnings subject to income taxes differs from the statutory federal rate for the years ended March 31, 2025 and 2024 due to the following:
| 2025 | 2024 | |||||||
| Income taxes (benefit) at federal statutory rate of 21.00% | $ | ( |
) | $ | ( |
) | ||
| Tax effect on non-deductible expenses and credits | ||||||||
| True up items | ( |
) | ||||||
| Expiration of net operating loss carryforwards (1) | ||||||||
| Change in valuation allowance | ||||||||
| Income Tax Expense (Benefit) | $ | $ | ||||||
______________
| (1) |
| F-23 |
ASC 740, “Income Taxes”, clarifies the accounting for uncertainty in income taxes recognized in an entity’s financial statements, and prescribes recognition thresholds and measurement attributes for financial statement disclosure of tax positions taken or expected to be taken on a tax return. Under ASC 740, the impact of an uncertain income tax position on the income tax return must be recognized at the largest amount that is more-likely-than-not to be sustained upon audit by the relevant taxing authority. An uncertain income tax position will not be recognized if it has less than a 50% likelihood of being sustained. Additionally, ASC 740 provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition. Our practice is to recognize interest and/or penalties related to income tax matters in income tax expense. During the years ended March 31, 2025 and 2024, we did not recognize any interest or penalties relating to tax matters.
At and for the years ended March 31, 2025 and
2024, management does not believe the Company has any uncertain tax positions. Accordingly, there are
Our tax returns remain open for examination by the applicable authorities, generally 3 years for federal and 4 years for state. We are currently not under examination by any taxing authorities.
8. COMMITMENTS AND CONTINGENCIES
CONTRACTUAL OBLIGATIONS AND COMMITMENTS
We have had the following material changes to our contractual obligations and commitments outside the ordinary course of business during the fiscal year ended March 31, 2025:
LEASE COMMITMENTS
Office, Lab and Manufacturing Space Leases
The office, lab and manufacturing leases are coterminous
with a remaining term of
As of March 31, 2025, we have right-of-use lease
assets of $
The following table presents a maturity analysis of expected undiscounted cash flows for operating leases on an annual basis for the next two fiscal years. All of our leases conterminously expire during the fiscal year ending March 31, 2027.
| Fiscal Years Ended March 31, | ||||
| 2026 | $ | |||
| 2027 | ||||
| Total minimum lease payments | ||||
| Less amount representing imputed interest | ( |
) | ||
| Present value of minimum lease payments | $ | |||
Overall, our rent expense, which is included in
general and administrative expenses, approximated $
| F-24 |
Premium Financing Agreement
In January 2025, the Company entered into a short-term premium financing
agreement with FIRST Insurance Funding, a division of Lake Forest Bank & Trust Company, N.A., to finance a portion of its Directors
& Officers (D&O) and other insurance premiums. The total amount financed under the agreement was approximately $
As collateral for the financing, the Company granted the lender a first priority security interest in the financed insurance policies, including all unearned premiums, dividends, credits, and certain loss payments. In the event of default, cancellation, or early termination of the policies, the lender has the right to collect any unearned premiums and apply them against the remaining loan balance.
This arrangement is classified as a short-term liability within other liabilities on the balance sheet (See Note 6) and is recorded net of any prepaid portions of the insurance policies.
LEGAL MATTERS
From time to time, claims are made against us in the ordinary course of business, which could result in litigation. Claims and associated litigation are subject to inherent uncertainties and unfavorable outcomes could occur, such as monetary damages, fines, penalties or injunctions prohibiting us from selling one or more products or engaging in other activities.
The occurrence of an unfavorable outcome in any specific period could have a material adverse effect on our results of operations for that period or future periods. We are not presently a party to any pending or threatened legal proceedings.
9. SEGMENT REPORTING
The Company operates as a single operating and reportable segment, which reflects the manner in which the Chief Operating Decision Maker (CODM), the Company’s Chief Executive Officer, manages the business and allocates resources. The Company is a development-stage medical technology company focused on advancing a clinical-stage therapeutic device, with key operational decisions based on cash availability, development milestones, and return on investment associated with future manufacturing and commercialization opportunities.
Although the Company has no commercial revenue, the CODM regularly reviews certain expense categories and cash flow metrics to assess progress and allocate resources. The primary internal measure of performance used by the CODM is cash used in operating activities, rather than traditional profit or loss measures.
In accordance with ASU 2023-07, which the Company adopted for the year ended March 31, 2025, the following significant expense categories and internal performance measures were reviewed by the CODM during the fiscal year ended March 31, 2025 and March 31, 2024:
| Category |
Year Ended March 31, 2025 |
Year Ended March 31, 2024 |
||||||
| Research and development1 | $ | $ | ||||||
| General and administrative2 | $ | $ | ||||||
| Cash used in operating activities3 | $ | $ | ||||||
| 1 | |
| 2 | |
| 3 |
| F-25 |
The Company does not allocate assets by segment, and the CODM does not use a measure of segment profit or loss to evaluate performance. There were no changes to the internal reports provided to or reviewed by the CODM during the years presented.
Entity-Wide Information:
| · | The Company did not recognize revenue during the fiscal year ended March 31, 2025. | |
| · | All long-lived assets are located in the United States. | |
| · | A significant portion of clinical trial activity is conducted through the Company’s wholly owned subsidiary based in Australia. |
10. SUBSEQUENT EVENTS
On June 25, 2025, the Company received notice from Nasdaq stating the Company has regained compliance with Listing Rule 5550(a)(2), and that the matter is now closed.
Reverse Split – Following the approval of a reverse stock split at a Special Meeting of Stockholders on May 13, 2025, our Board of Directors approved a 1-for-8 reverse stock split or our outstanding shares of Common Stock, effective as of the close of business on June 6, 2025 with an effective trading date of June 9, 2025. Accordingly, each 8 shares of outstanding common stock held by stockholders were combined into one share of common stock. Our authorized common stock remained at 60,000,000 shares following the stock split. We issued an additional 77 shares as a result of rounding up fractional shares related to the reverse stock split.
On June 2 and June 16, 2025, the second and third patients, respectively, were treated with the Aethlon Hemopurifier at GenesisCare North Shore in Sydney, Australia. Each patient underwent a single 4-hour treatment session and tolerated the procedure without complications. Follow-up visits will include safety assessments, extracellular vesicle (EV) and T cell measurements, as well as imaging to evaluate clinical response.
RSU Grants
In April 2025, our Board of Directors approved, pursuant to the terms of the Director Compensation Policy, the grant of the annual RSUs under the Director Compensation Policy to each of the four non-employee directors of the Company then serving on the Board of Directors. The Director Compensation Policy provides for a grant of stock options or $50,000 worth of RSUs at the beginning of each fiscal year for current non-employee directors then serving on the Board of Directors, and for a grant of stock options or $75,000 worth of RSUs for a newly elected non-employee director, with each RSU priced at the average for the closing prices for the five days preceding and including the date of grant, or $2.80 per share for the RSUs granted in April 2025. As a result, in April 2025 the four eligible directors were each granted an RSU in the amount of 17,858 shares under the Company’s 2020 Equity Incentive Plan, or the 2020 Plan. The RSUs are subject to vesting in four equal installments, with 25% of the restricted stock units vesting on each of June 30, 2025, September 30, 2025, December 31, 2025, and March 31, 2026, subject in each case to the director’s Continuous Service (as defined in the 2020 Plan), through such dates. Vesting will terminate upon the director’s termination of Continuous Service prior to any vesting date.
| F-26 |